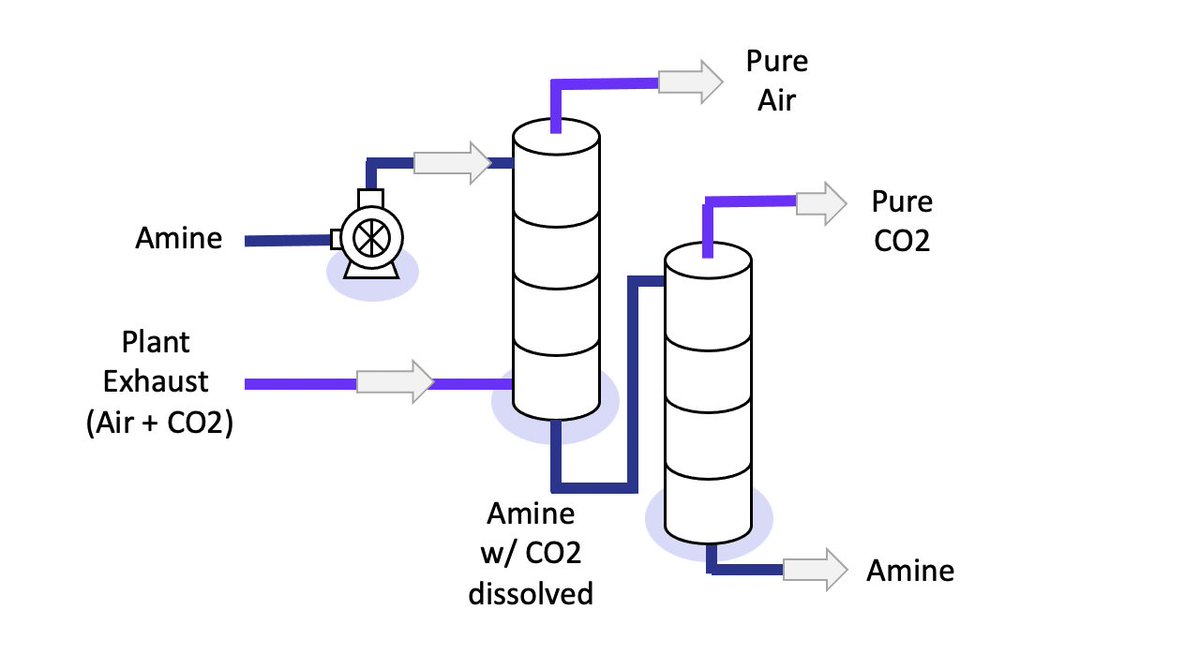
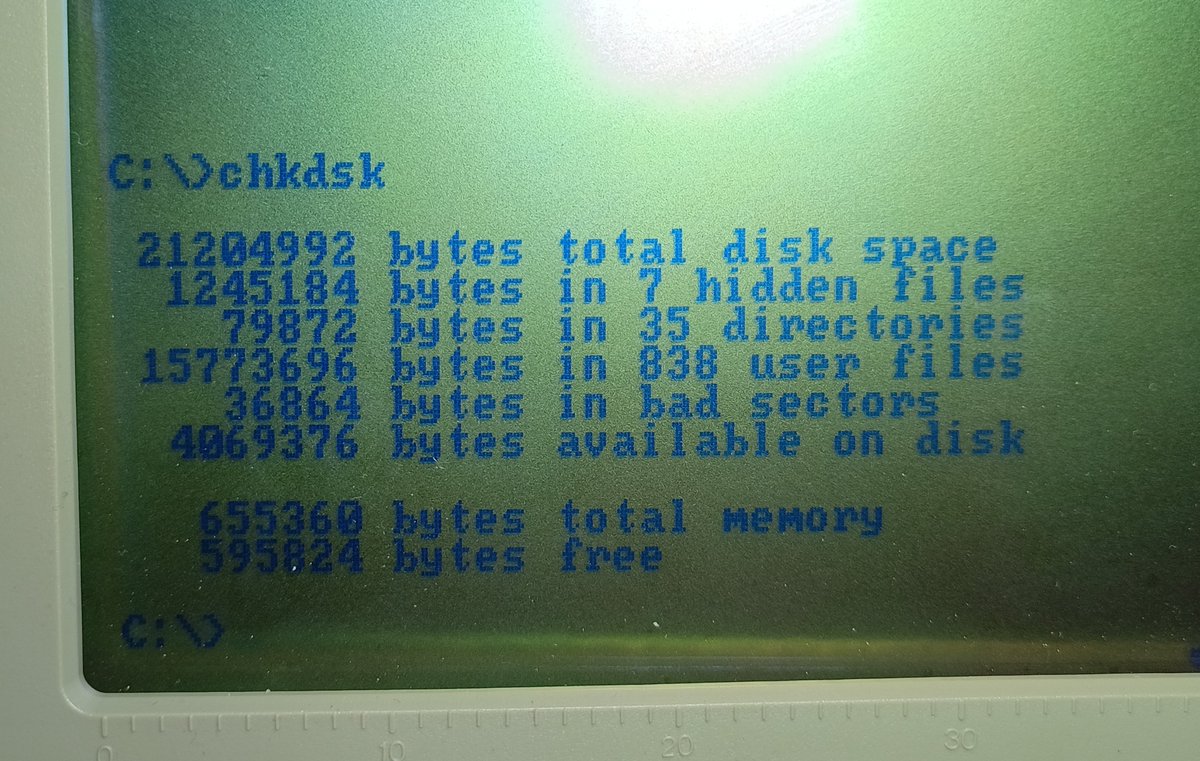
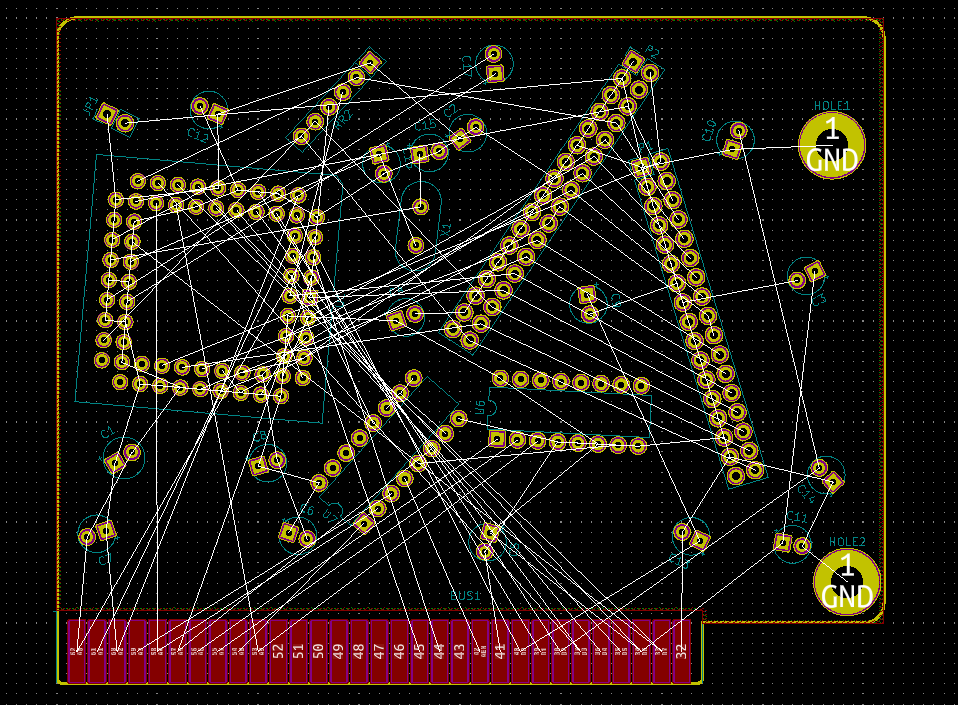
I think I've permanently confused KiCAD
it's reminding me that I need to connect this capacitor to Nothing
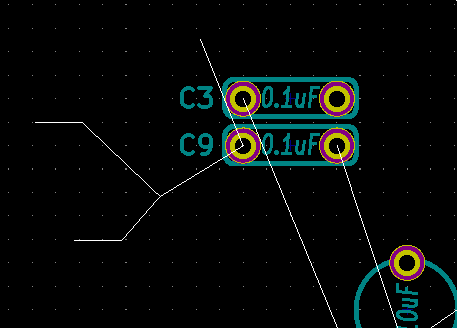


they have an ISA footprint but it doesn't show up when you search for "ISA" because they called it BUS_AT despite it being clearly called "ISA" since THE LATE 80s

The full AT bus, the 16bit extension to the 8bit PC/XT bus!






this will fix and/or cause all my problems
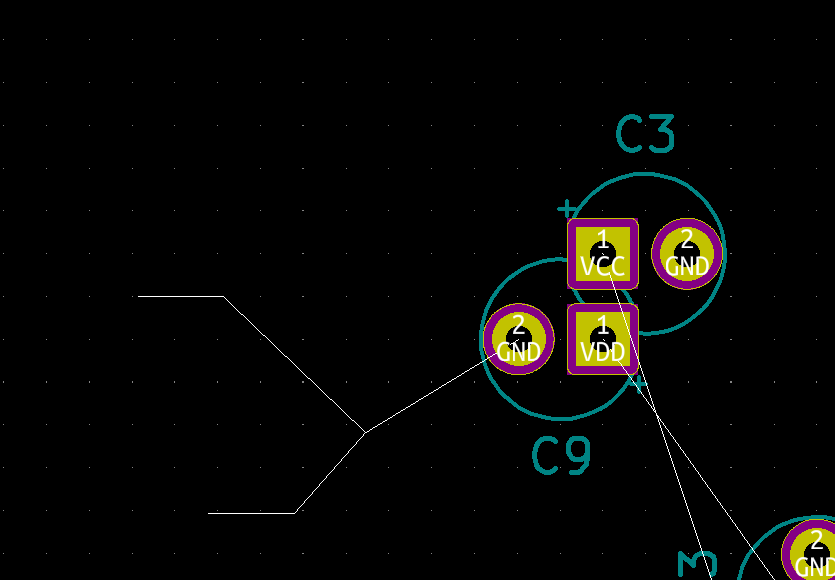
look at this.
it's the wrong 3D library, as that's just the footprint not the socket. I don't even know if here's a socket, so I'd like to look.
See the 3D model path? it's in Package_LCC.3dshapes


1. the source for kicad which points at this non-existent file
2. @TubeTimeUS's PlaidBib project which points at "Housings_LCC.3dshapes/PLCC-68_THT-Socket.wrl", which is a slightly different path!

https://t.co/6pHY6xZkCZ
Drinks available:
— foone (@Foone) March 24, 2019
Sprite
Diet Coke
\uff34 \uff28 \uff25 \uff36 \uff2f \uff29 \uff24 pic.twitter.com/t2FXeaJAyy
why
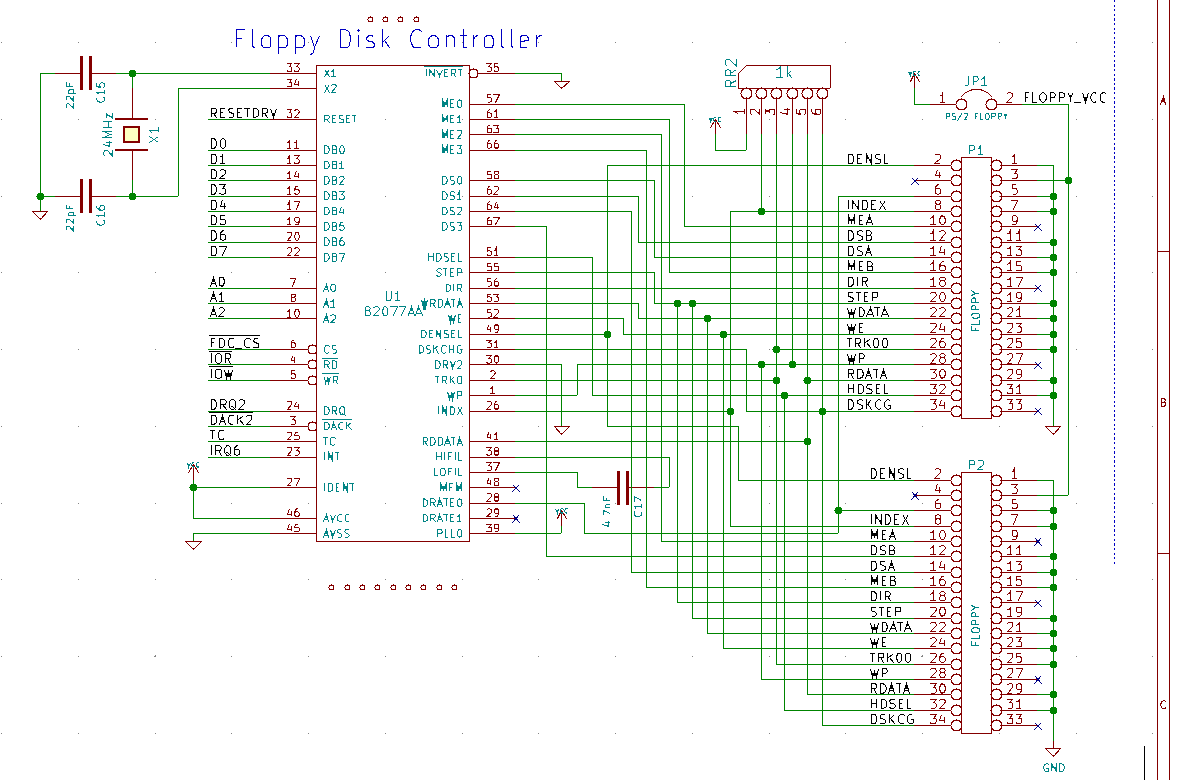
TWO HOURS LATER I'M DESIGNING A FLOPPY DISK CONTROLLER


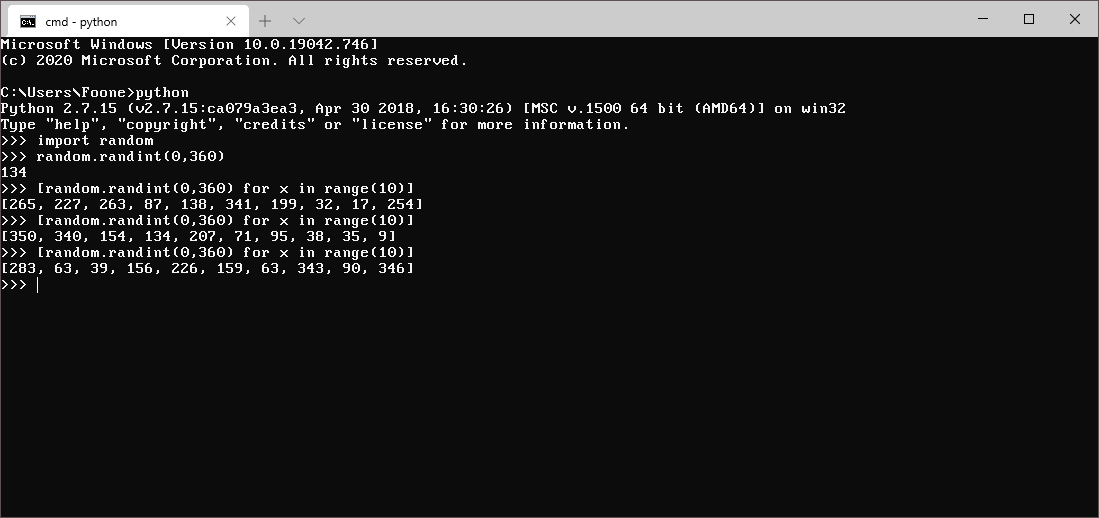
Obviously I let a computer generate the randomness, I'm not a barbarian.

More from foone









A fun fact on the wikipedia page for the metal–oxide–semiconductor field-effect transistor:
it is the most frequently manufactured device in history, and the total number manufactured from 1960-2018 is 13 sextillion.
That's 13,000,000,000,000,000,000,000.

Though this picture is a bit misleading.
Even with devices this small, we couldn't make 13 sextillion of them in 60 years.
So imagine a chip like this. It's the 555 timer, which is one of the most popular integrated circuits ever made.
In 2017, it was estimated a billion are made every year.

And at the heart of it is the die, which looks like this:
(from Ken Shirriff's blog)
https://t.co/mz5PQDjYqF
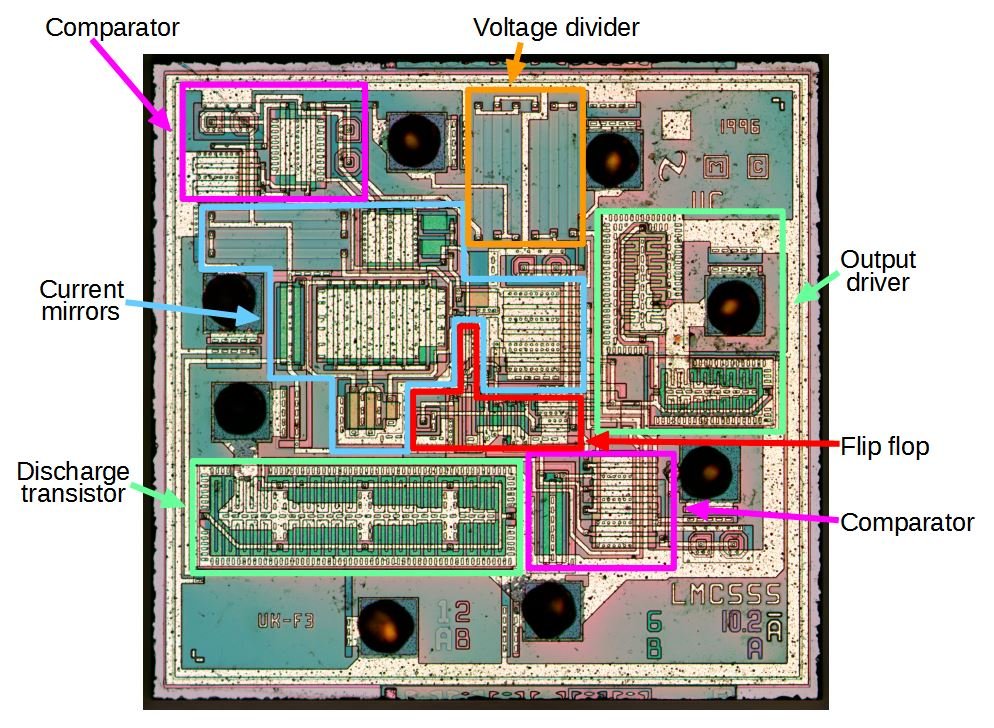
And that's fundamentally a bunch of CMOS transistors (along with some diodes and resistors), which are a type of MOSFET. How many of them are on a 555?
about 25. Not many, but it's a very simple chip.
it is the most frequently manufactured device in history, and the total number manufactured from 1960-2018 is 13 sextillion.
That's 13,000,000,000,000,000,000,000.
Though this picture is a bit misleading.
Even with devices this small, we couldn't make 13 sextillion of them in 60 years.
So imagine a chip like this. It's the 555 timer, which is one of the most popular integrated circuits ever made.
In 2017, it was estimated a billion are made every year.
And at the heart of it is the die, which looks like this:
(from Ken Shirriff's blog)
https://t.co/mz5PQDjYqF
And that's fundamentally a bunch of CMOS transistors (along with some diodes and resistors), which are a type of MOSFET. How many of them are on a 555?
about 25. Not many, but it's a very simple chip.
More from Science

https://t.co/hXlo8qgkD0
Look like that they got a classical case of PCR Cross-Contamination.
They had 2 fabricated samples (SRX9714436 and SRX9714921) on the same PCR run. Alongside with Lung07. They did not perform metagenomic sequencing on the “feces” and they did not get

A positive oral or anal swab from anywhere in their sampling. Feces came from anus and if these were positive the anal swabs must also be positive. Clearly it got there after the NA have been extracted and were from the very low-level degraded RNA which were mutagenized from
The Taq. https://t.co/yKXCgiT29w to see SRX9714921 and SRX9714436.
Human+Mouse in the positive SRA, human in both of them. Seeing human+mouse in identical proportions across 3 different sequencers (PRJNA573298, A22, SEX9714436) are pretty straight indication that the originals
Were already contaminated with Human and mouse from the very beginning, and that this contamination is due to dishonesty in the sample handling process which prescribe a spiking of samples in ACE2-HEK293T/A549, VERO E6 and Human lung xenograft mouse.
The “lineages” they claimed to have found aren’t mutational lineages at all—all the mutations they see on these sequences were unique to that specific sequence, and are the result of RNA degradation and from the Taq polymerase errors accumulated from the nested PCR process

Look like that they got a classical case of PCR Cross-Contamination.
They had 2 fabricated samples (SRX9714436 and SRX9714921) on the same PCR run. Alongside with Lung07. They did not perform metagenomic sequencing on the “feces” and they did not get
A positive oral or anal swab from anywhere in their sampling. Feces came from anus and if these were positive the anal swabs must also be positive. Clearly it got there after the NA have been extracted and were from the very low-level degraded RNA which were mutagenized from
The Taq. https://t.co/yKXCgiT29w to see SRX9714921 and SRX9714436.
Human+Mouse in the positive SRA, human in both of them. Seeing human+mouse in identical proportions across 3 different sequencers (PRJNA573298, A22, SEX9714436) are pretty straight indication that the originals
Were already contaminated with Human and mouse from the very beginning, and that this contamination is due to dishonesty in the sample handling process which prescribe a spiking of samples in ACE2-HEK293T/A549, VERO E6 and Human lung xenograft mouse.
The “lineages” they claimed to have found aren’t mutational lineages at all—all the mutations they see on these sequences were unique to that specific sequence, and are the result of RNA degradation and from the Taq polymerase errors accumulated from the nested PCR process




All modern research questions frame your mindset and self-frame research paradigm. Broad thinking: how little of everything can a citizen survive on; how cheap can your upkeep be? /1
When an American patient lands in an Austrian hospital for a back problem, a doctor tells him to perform a set of exercises.
- How many?
- Do you have anything else to do? /2
This interchange illustrates two mindsets colliding at bedside. How little can I get away with vs there is no limit to effort when it comes to your wellness. /3
When you were robbed of movement, somebody started selling you exercise. To understand that digging a ditch, to build a house, or to carry a child around, or waking to your grandparents for an hour is not the same as jogging on a treadmill... will reveal what research hides.
/4
When I talk about doing a purposeful activity outdoors, I look at complexity of movement, purpose, meaning, sun, and air, even an opportunity to meet a neighbor... that is now reduced to a calcium pill, vitamin D, an antidepressant, an osteoporosis shot, and an oxygen tank. /5
Is moderate exercise enough to live as long as possible, or should you be doing vigorous exercise? And what proportion is best? This article has the answers. https://t.co/YJqpaaI0UR
— Sebastian Rushworth M.D. (@sebrushworth) January 24, 2021
When an American patient lands in an Austrian hospital for a back problem, a doctor tells him to perform a set of exercises.
- How many?
- Do you have anything else to do? /2
This interchange illustrates two mindsets colliding at bedside. How little can I get away with vs there is no limit to effort when it comes to your wellness. /3
When you were robbed of movement, somebody started selling you exercise. To understand that digging a ditch, to build a house, or to carry a child around, or waking to your grandparents for an hour is not the same as jogging on a treadmill... will reveal what research hides.
/4
When I talk about doing a purposeful activity outdoors, I look at complexity of movement, purpose, meaning, sun, and air, even an opportunity to meet a neighbor... that is now reduced to a calcium pill, vitamin D, an antidepressant, an osteoporosis shot, and an oxygen tank. /5