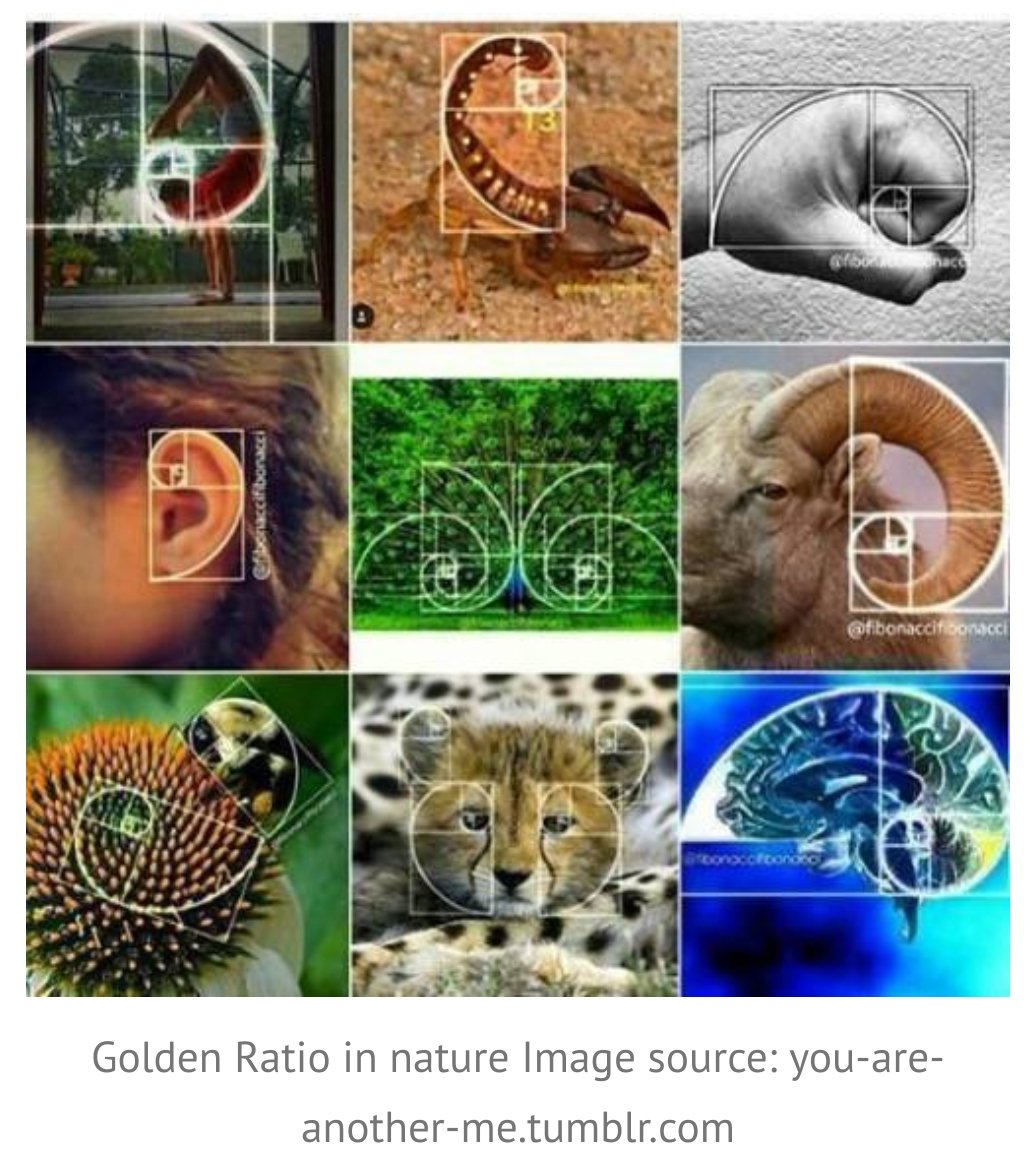
Hard agree. And if this is useful, let me share something that often gets omitted (not by @kakape).
Variants always emerge, & are not good or bad, but expected. The challenge is figuring out which variants are bad, and that can't be done with sequence alone.
Feels like the next thing we're going to need is a ranking system for how concerning "variants of concern\u201d actually are.
— Kai Kupferschmidt (@kakape) January 15, 2021
A lot of constellations of mutations are concerning, but people are lumping together variants with vastly different levels of evidence that we need to worry.
-More viral shedding (infected people produce more virus)
-Increased interval of viral shedding (people are contagious for longer)
-Increased viral fitness (virus can replicate better in infected people)
-Better receptor binding (virus can infect cells more easily)
-Increased environmental stability (virus can remain infectious in the environment for longer)
-Increased ability to evade host defenses (virus can escape innate cellular defenses against infection and thus replicate better)
Could be one, some, or all of the above
-Render masks or other physical barriers useless
-Make the virus transmissible by a totally different route of infection
-Turn the virus into the infectious equivalent of a smart cruise missile
-Defy the laws of conservation of mass and energy
More from Science

Hugh Everett's birthday! Pioneer of the Many-Worlds Interpretation of quantum mechanics. Let us celebrate by thinking about ontological extravagance. I will do so by way of analogy, because I have found that everyone loves analogies and nobody ever willfully misconstrues them.
We look at the night sky and see photons arriving to us, emitted by distant stars. Let's contrast two different theories about how stars emit photons.
One theory says, we know how stars shine, and our equations predict that they emit photons roughly uniformly in all directions. Call this the "Many-Photons Interpretation" (MPI).
But! Others object. That is *so many photons*. Most of which we don't observe, and can't observe, since they're moving away at the speed of light. It's too ontologically extravagant to posit a huge number of unobservable things!
So they suggest a "Photon Collapse Interpretation." According to this theory, the photons emitted toward us actually exist. But photons that would be emitted in directions we will never observe simply collapse into utter non-existence.
The physicist Hugh Everett III was born #OTD in 1930. His \u201crelative state\u201d formulation of quantum mechanics, which we now call the \u201cMany Worlds Interpretation,\u201d was published in 1957. pic.twitter.com/ZqMsZcPJDG
— Robert McNees, the bastegod (@mcnees) November 11, 2020
We look at the night sky and see photons arriving to us, emitted by distant stars. Let's contrast two different theories about how stars emit photons.
One theory says, we know how stars shine, and our equations predict that they emit photons roughly uniformly in all directions. Call this the "Many-Photons Interpretation" (MPI).
But! Others object. That is *so many photons*. Most of which we don't observe, and can't observe, since they're moving away at the speed of light. It's too ontologically extravagant to posit a huge number of unobservable things!
So they suggest a "Photon Collapse Interpretation." According to this theory, the photons emitted toward us actually exist. But photons that would be emitted in directions we will never observe simply collapse into utter non-existence.



Recently I learned something about DNA that blew my mind, and in this thread, I'll attempt to blow your mind as well. Behold: Chargaff's 2nd Parity Rule for DNA N-Grams.
If you are into cryptography or reverse engineering, you should love this.
Thread:

DNA consists of four different 'bases', A, C, G and T. These bases have specific meaning within our biology. Specifically, within the 'coding part' of a gene, a triplet of bases encodes for an amino acid

Most DNA is stored redundantly, in two connected strands. Wherever there is an A on one strand, you'll find a T on the other one. And similarly for C and G:
T G T C A G T
A C A G T C A
(note how the other strand is upside down - this matters!)

If you take all the DNA of an organism (both strands), you will find equal numbers of A's and T's, as well as equal numbers of C's and G's. This is true by definition.
This is called Chargaff's 1st parity rule.
https://t.co/jD4cMt0PJ0

Strangely enough, this rule also holds per strand! So even if you take away the redundancy, there are 99% equal numbers of A/T and C/G * on each strand *. And we don't really know why.
This is called Chargaff's 2nd parity rule.

If you are into cryptography or reverse engineering, you should love this.
Thread:
DNA consists of four different 'bases', A, C, G and T. These bases have specific meaning within our biology. Specifically, within the 'coding part' of a gene, a triplet of bases encodes for an amino acid
Most DNA is stored redundantly, in two connected strands. Wherever there is an A on one strand, you'll find a T on the other one. And similarly for C and G:
T G T C A G T
A C A G T C A
(note how the other strand is upside down - this matters!)
If you take all the DNA of an organism (both strands), you will find equal numbers of A's and T's, as well as equal numbers of C's and G's. This is true by definition.
This is called Chargaff's 1st parity rule.
https://t.co/jD4cMt0PJ0
Strangely enough, this rule also holds per strand! So even if you take away the redundancy, there are 99% equal numbers of A/T and C/G * on each strand *. And we don't really know why.
This is called Chargaff's 2nd parity rule.