
Let Uncle Ralph educate you
Synthetic Viral Genomics:
Risks and Benefits for Science and Society
These enzymes can be used to create unique interconnecting junctions, which can be subsequently removed from final assembly product allowing seamless reconstruction of an exact sequence

The pathogenicity of these chimeric coronaviruses is unknown
The pathogenicity of these chimeric coronaviruses is unknown
The pathogenicity of these chimeric coronaviruses is unknown
This powerful technique provides bioterrorists with a “scapegoat” option; leaving a sequence signature that misdirects efforts at tracking the true originators of the crime. Even better, the approach could be used to build mistrust &/or precipitate open warfare



Hence Ecohealth DARPA/DTRA spooks & virus thieves collaboration with Baric (UNC) Lipkin (Mailman) NIchols (Atlanta CDC) and USAMRIID (Bavari, Totura et al) & Jonathan Epstein's palpable concern about dual use references in the @USRightToKnow FOIA emails

Delving into the cold and calculating mind of a twisted genius?
https://t.co/MrwxYK93cx
Bring Uncle Ralph and his transgenic mice in for questioning!
unroll @threadreaderapp

https://t.co/rjNZ8aDNDl

1. Synthetic Genomics: Options for Governance (2008) https://t.co/A32jEfL1TQ
2. Sequence Screening - Robert Jones (2005)
https://t.co/zOqMroYbVU
3. Synthetic Biology as a Field of Dual-Use Bioethical Concern - Alexander Kelle
https://t.co/Va1movPqvs
4. Sanghvi Y. A Roadmap to the Assembly of Synthetic DNA from Raw Materials.
https://t.co/4kWu4vzcXC
5. Collett MS. Impact of Synthetic Genomics on the Threat of Bioterrorism with Viral Agents.
https://t.co/OcaTmuCUBy
6. Fleming DO. Risk Assessment of Synthetic Genomics: A Biosafety & Biosecurity Perspective.
https://t.co/JBcLVLr005
7. Risk Governance of Synthetic Biology
https://t.co/9BE9X5s0Fb
8. US Competitiveness in Synthetic Biology
https://t.co/OCZJdsXSjl
9. Ensuring security of synthetic biology
https://t.co/7kPE1OpbrS
10. Synthetic biology: emerging research field in China
https://t.co/T3BcaDvUsa
11.
What rough beast? Synthetic biology, uncertainty,& the future of biosecurity (2016)
https://t.co/XTlBXILEWh

Reverse genetics with a full-length infectious cDNA of severe acute respiratory syndrome coronavirus (2003)
https://t.co/k8PI1rbcm8
Systematic Assembly of a Full-Length Infectious Clone of Human Coronavirus NL63 (2008)
https://t.co/bWhAv1wsC0
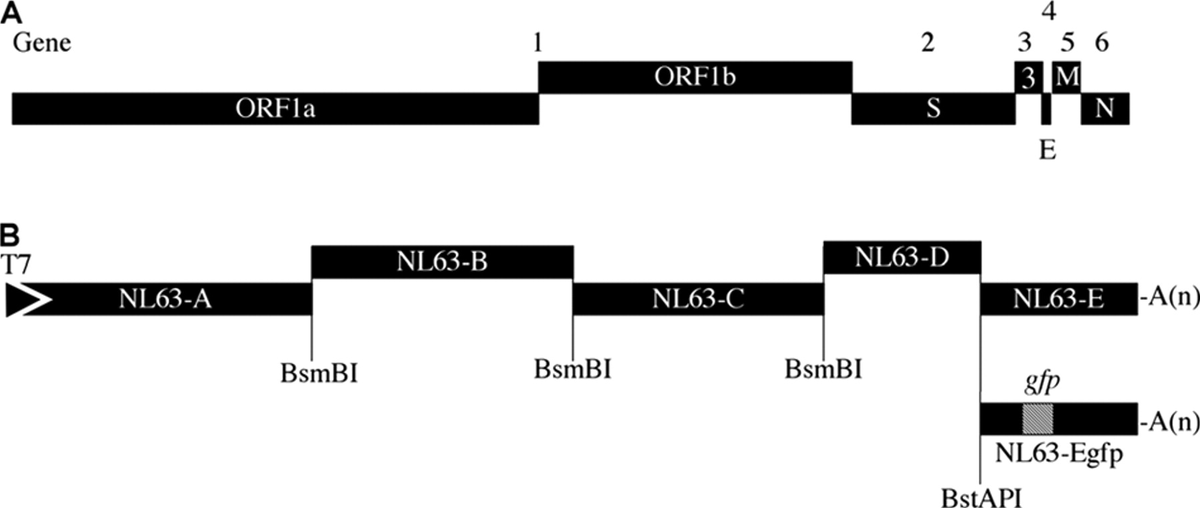
In 2016, Shi and her team at the WIV, in conjunction with the New York-based EcoHealth Alliance, constructed a full-length clone of a bat coronavirus called SL-CoV WIV1. They assembled it in discrete segments.
“Thus, several options exist to remove the desired insert DNA with a single restriction digestion.”
This shows that researchers at the WIV have the ability to genetically engineer viruses and remove the signatures of the genetic engineering.
They showed how they can insert new spikes into viruses. The researchers state:
“Then any spike could be substituted into the genome of SARSr-CoV WIV1 through this strategy.”
COVID 19: The Spike and the Furin Cleavage
https://t.co/bDOMmhpJ1t
More from Billy Bostickson 🏴👁&👁 🆓




Keep dwelling on this:
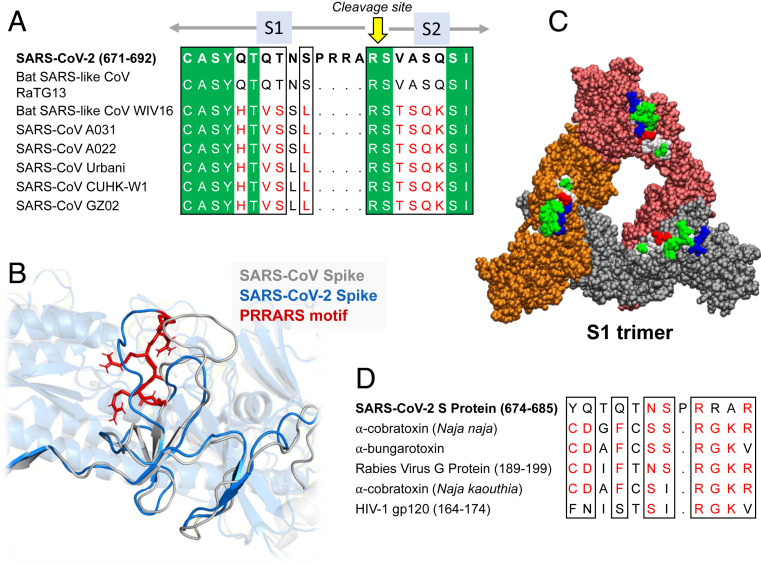
Further Examination of the Motif near PRRA Reveals Close Structural Similarity to the SEB Superantigen as well as Sequence Similarities to Neurotoxins and a Viral SAg.
The insertion PRRA together with 7 sequentially preceding residues & succeeding R685 (conserved in β-CoVs) form a motif, Y674QTQTNSPRRAR685, homologous to those of neurotoxins from Ophiophagus (cobra) and Bungarus genera, as well as neurotoxin-like regions from three RABV strains
(20) (Fig. 2D). We further noticed that the same segment bears close similarity to the HIV-1 glycoprotein gp120 SAg motif F164 to V174.
https://t.co/EwwJOSa8RK
In (B), the segment S680PPRAR685 including the PRRA insert and highly conserved cleavage site *R685* is shown in van der Waals representation (black labels) and nearby CDR residues of the TCRVβ domain are labeled in blue/white
https://t.co/BsY8BAIzDa
Sequence Identity %
https://t.co/BsY8BAIzDa
Y674 - QTQTNSPRRA - R685
Similar to neurotoxins from Ophiophagus (cobra) & Bungarus genera & neurotoxin-like regions from three RABV strains
T678 - NSPRRA- R685
Superantigenic core, consistently aligned against bacterial or viral SAgs

Further Examination of the Motif near PRRA Reveals Close Structural Similarity to the SEB Superantigen as well as Sequence Similarities to Neurotoxins and a Viral SAg.
The insertion PRRA together with 7 sequentially preceding residues & succeeding R685 (conserved in β-CoVs) form a motif, Y674QTQTNSPRRAR685, homologous to those of neurotoxins from Ophiophagus (cobra) and Bungarus genera, as well as neurotoxin-like regions from three RABV strains
(20) (Fig. 2D). We further noticed that the same segment bears close similarity to the HIV-1 glycoprotein gp120 SAg motif F164 to V174.
https://t.co/EwwJOSa8RK
In (B), the segment S680PPRAR685 including the PRRA insert and highly conserved cleavage site *R685* is shown in van der Waals representation (black labels) and nearby CDR residues of the TCRVβ domain are labeled in blue/white
https://t.co/BsY8BAIzDa
Sequence Identity %
https://t.co/BsY8BAIzDa
Y674 - QTQTNSPRRA - R685
Similar to neurotoxins from Ophiophagus (cobra) & Bungarus genera & neurotoxin-like regions from three RABV strains
T678 - NSPRRA- R685
Superantigenic core, consistently aligned against bacterial or viral SAgs
1/x Fort Detrick History
Mr. Patrick, one of the chief scientists at the Army Biological Warfare Laboratories at Fort Detrick in Frederick, Md., held five classified US patents for the process of weaponizing anthrax.
2/x
Under Mr. Patrick’s direction, scientists at Fort Detrick developed a tularemia agent that, if disseminated by airplane, could cause casualties & sickness over 1000s mi². In a 10,000 mi² range, it had 90% casualty rate & 50% fatality rate

3/x His team explored Q fever, plague, & Venezuelan equine encephalitis, testing more than 20 anthrax strains to discern most lethal variety. Fort Detrick scientists used aerosol spray systems inside fountain pens, walking sticks, light bulbs, & even in 1953 Mercury exhaust pipes

4/x After retiring in 1986, Mr. Patrick remained one of the world’s foremost specialists on biological warfare & was a consultant to the CIA, FBI, & US military. He debriefed Soviet defector Ken Alibek, the deputy chief of the Soviet biowarfare program
https://t.co/sHqSaTSqtB

5/x Back in Time
In 1949 the Army created a small team of chemists at "Camp Detrick" called Special Operations Division. Its assignment was to find military uses for toxic bacteria. The coercive use of toxins was a new field, which fascinated Allen Dulles, later head of the CIA

Mr. Patrick, one of the chief scientists at the Army Biological Warfare Laboratories at Fort Detrick in Frederick, Md., held five classified US patents for the process of weaponizing anthrax.
2/x
Under Mr. Patrick’s direction, scientists at Fort Detrick developed a tularemia agent that, if disseminated by airplane, could cause casualties & sickness over 1000s mi². In a 10,000 mi² range, it had 90% casualty rate & 50% fatality rate
3/x His team explored Q fever, plague, & Venezuelan equine encephalitis, testing more than 20 anthrax strains to discern most lethal variety. Fort Detrick scientists used aerosol spray systems inside fountain pens, walking sticks, light bulbs, & even in 1953 Mercury exhaust pipes
4/x After retiring in 1986, Mr. Patrick remained one of the world’s foremost specialists on biological warfare & was a consultant to the CIA, FBI, & US military. He debriefed Soviet defector Ken Alibek, the deputy chief of the Soviet biowarfare program
https://t.co/sHqSaTSqtB
5/x Back in Time
In 1949 the Army created a small team of chemists at "Camp Detrick" called Special Operations Division. Its assignment was to find military uses for toxic bacteria. The coercive use of toxins was a new field, which fascinated Allen Dulles, later head of the CIA
1. Strange goings on down at Uncle Sam's Farm in Ukraine?
h/t @DrKevinWMcCair1 video:
https://t.co/1lrFeWcakB
From Daily Expose:
U.S. DoD awarded a contract for ‘COVID-19 Research’ in Ukraine 3 months before Covid was known to even
2. The findings seem to be kosher and can "currently" be confirmed by:
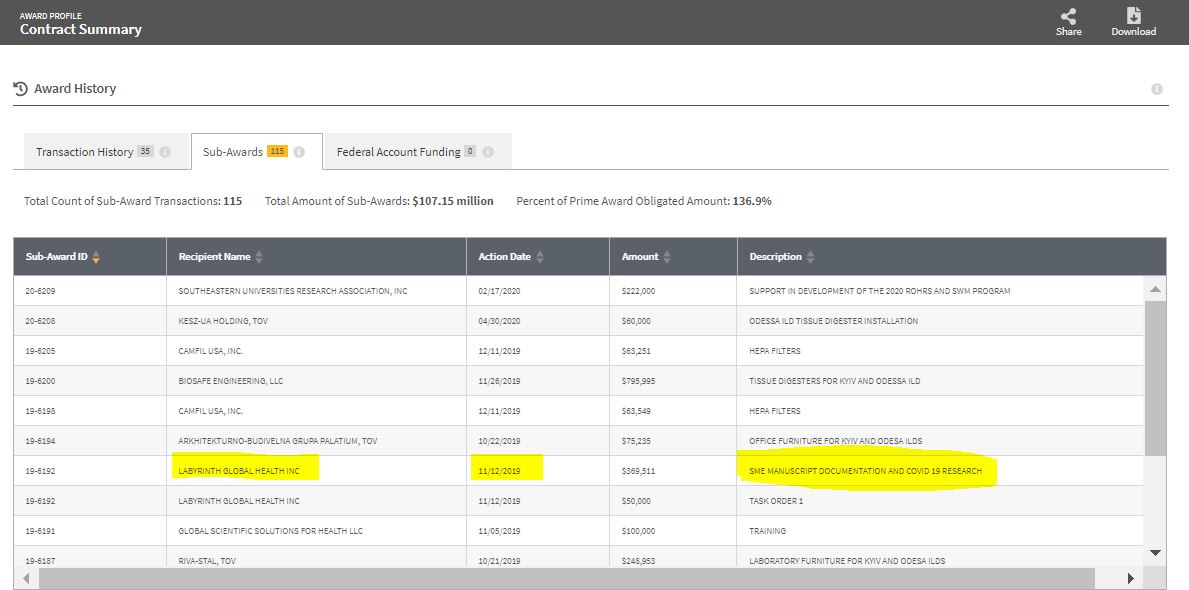
1. checking the DOD Award to Black & Veatch
CONT_IDV_HDTRA108D0007_9700, here:
https://t.co/G5ZEUhPm5U
2. click the tab for sub-awards
3. 7th down from top
4. 19-6192 is the sub-award ID
5. Note Date
3. Download the CSV file by clicking top right:
Again Data is confirmed (with more detail in Column 32 in the csv file)
4. CSV Details (Column 32)
Note Kiev, Ukraine Mentioned and Date 2019
5. CSV Details (2)
Mentions Labyrinth's HQ in Saint Petersburg, Florida, not Russia ;)
URL in CSV directs to original URL
https://t.co/G5ZEUhPm5U
Copy & Paste URL, not click.
Last column with date does not refute finding, it is:
"subaward_fsrs_report_last_modified_date"

h/t @DrKevinWMcCair1 video:
https://t.co/1lrFeWcakB
From Daily Expose:
U.S. DoD awarded a contract for ‘COVID-19 Research’ in Ukraine 3 months before Covid was known to even
2. The findings seem to be kosher and can "currently" be confirmed by:
1. checking the DOD Award to Black & Veatch
CONT_IDV_HDTRA108D0007_9700, here:
https://t.co/G5ZEUhPm5U
2. click the tab for sub-awards
3. 7th down from top
4. 19-6192 is the sub-award ID
5. Note Date
3. Download the CSV file by clicking top right:
Again Data is confirmed (with more detail in Column 32 in the csv file)
4. CSV Details (Column 32)
Note Kiev, Ukraine Mentioned and Date 2019
5. CSV Details (2)
Mentions Labyrinth's HQ in Saint Petersburg, Florida, not Russia ;)
URL in CSV directs to original URL
https://t.co/G5ZEUhPm5U
Copy & Paste URL, not click.
Last column with date does not refute finding, it is:
"subaward_fsrs_report_last_modified_date"
More from Science

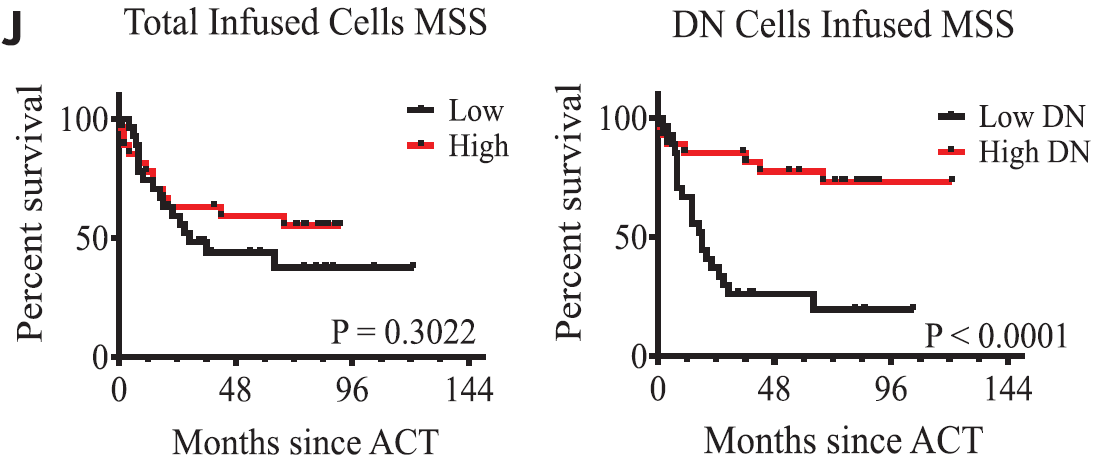
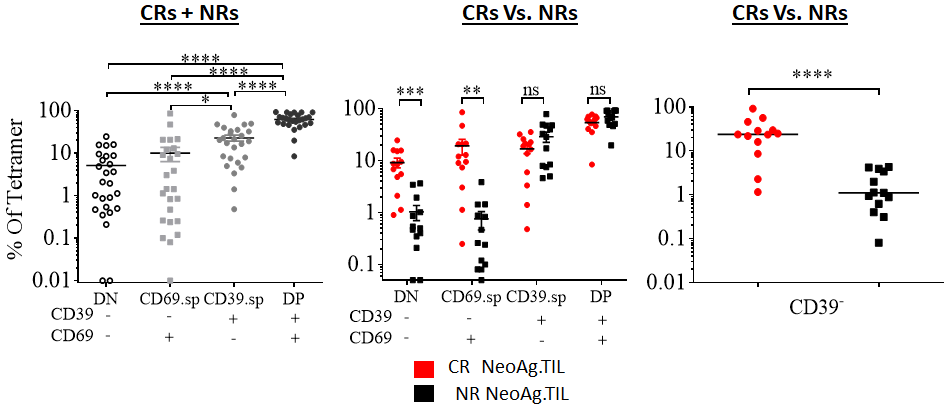

💥and so it begins..💥
It's time, my friends 🤩🤩
[Thread] #ProjectOdin
https://t.co/fO90N78fta
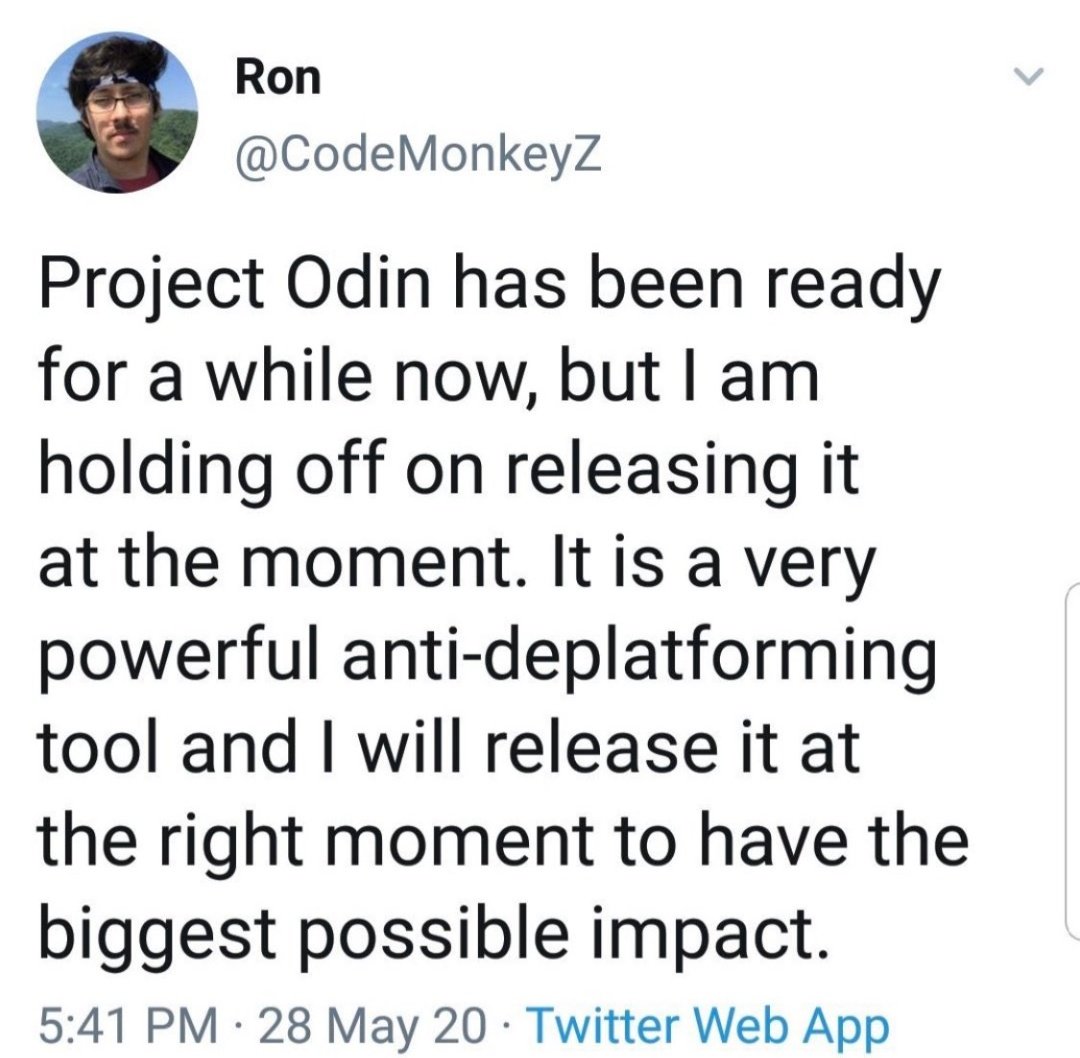
new quantum-based internet #ElonMusk #QVS #QFS
Political justification ⏬⏬
#ProjectOdin
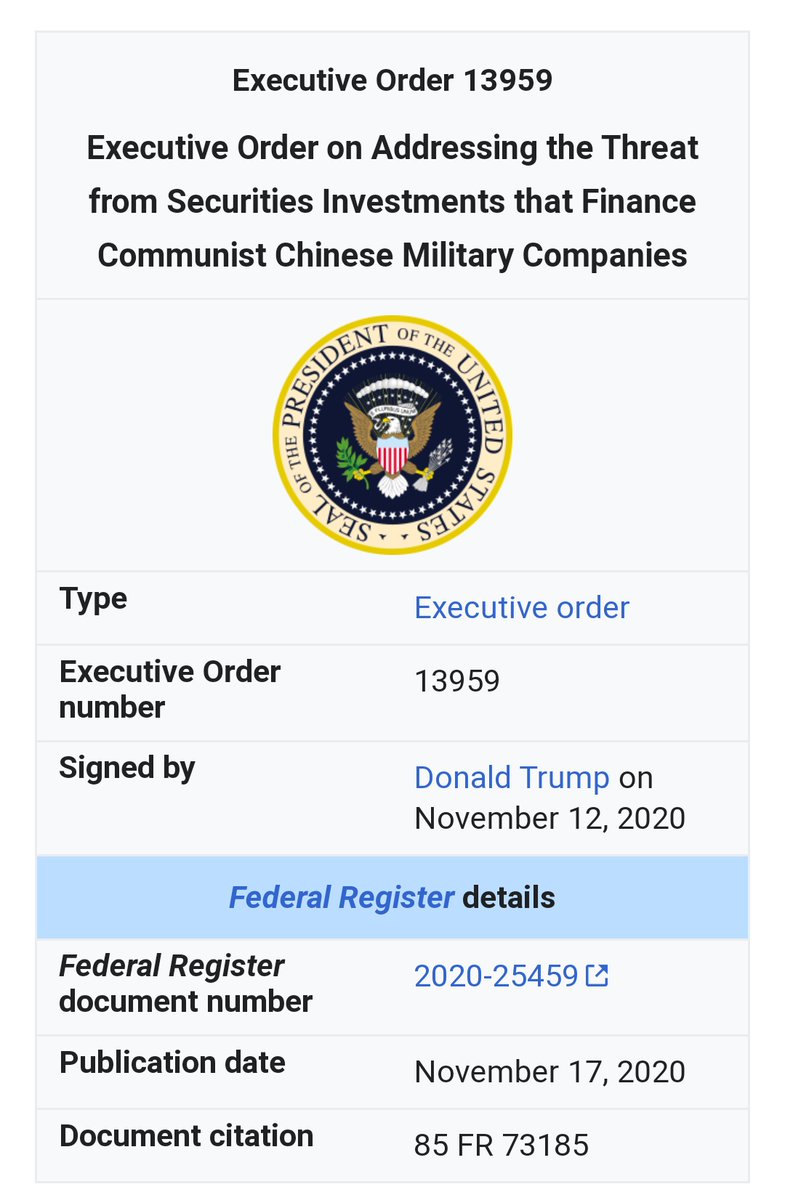
#ProjectOdin #Starlink #ElonMusk #QuantumInternet
It's time, my friends 🤩🤩
[Thread] #ProjectOdin
The Alliance has Project Odin ready to go - the new quantum-based internet. #ElonMusk #QVS #QFS #ProjectOdin
— Der Preu\xdfe Parler: @DerPreusse (@DerPreusse1963) January 12, 2021
https://t.co/fO90N78fta
new quantum-based internet #ElonMusk #QVS #QFS
Political justification ⏬⏬
#ProjectOdin
#ProjectOdin #Starlink #ElonMusk #QuantumInternet


Ever since @JesseJenkins and colleagues work on a zero carbon US and this work by @DrChrisClack and colleagues on incorporating DER, I've been having the following set of thoughts about how to reduce the risk of failure in a US clean energy buildout. Bottom line is much more DER.
Typically, when we see zero-carbon electricity coupled to electrification of transport and buildings, implicitly standing behind that is totally unprecedented buildout of the transmission system. The team from Princeton's modeling work has this in spades for example.
But that, more even than the new generation required, runs straight into a thicket/woodchipper of environmental laws and public objections that currently (and for the last 50y) limit new transmission in the US. We built most transmission prior to the advent of environmental law.
So what these studies are really (implicitly) saying is that NEPA, CEQA, ESA, §404 permitting, eminent domain law, etc, - and the public and democratic objections that drive them - will have to change in order to accommodate the necessary transmission buildout.
I live in a D supermajority state that has, for at least the last 20 years, been in the midst of a housing crisis that creates punishing impacts for people's lives in the here-and-now and is arguably mostly caused by the same issues that create the transmission bottlenecks.
Rooftop solar can play a key role in a transition to 100% renewable energy - and it can help American's pocketbooks #GoSolarhttps://t.co/6p9jb62EGW
— Environment America (@EnvAm) January 14, 2021
Typically, when we see zero-carbon electricity coupled to electrification of transport and buildings, implicitly standing behind that is totally unprecedented buildout of the transmission system. The team from Princeton's modeling work has this in spades for example.
But that, more even than the new generation required, runs straight into a thicket/woodchipper of environmental laws and public objections that currently (and for the last 50y) limit new transmission in the US. We built most transmission prior to the advent of environmental law.
So what these studies are really (implicitly) saying is that NEPA, CEQA, ESA, §404 permitting, eminent domain law, etc, - and the public and democratic objections that drive them - will have to change in order to accommodate the necessary transmission buildout.
I live in a D supermajority state that has, for at least the last 20 years, been in the midst of a housing crisis that creates punishing impacts for people's lives in the here-and-now and is arguably mostly caused by the same issues that create the transmission bottlenecks.



You May Also Like












"I really want to break into Product Management"
make products.
"If only someone would tell me how I can get a startup to notice me."
Make Products.
"I guess it's impossible and I'll never break into the industry."
MAKE PRODUCTS.
Courtesy of @edbrisson's wonderful thread on breaking into comics – https://t.co/TgNblNSCBj – here is why the same applies to Product Management, too.
There is no better way of learning the craft of product, or proving your potential to employers, than just doing it.
You do not need anybody's permission. We don't have diplomas, nor doctorates. We can barely agree on a single standard of what a Product Manager is supposed to do.
But – there is at least one blindingly obvious industry consensus – a Product Manager makes Products.
And they don't need to be kept at the exact right temperature, given endless resource, or carefully protected in order to do this.
They find their own way.
make products.
"If only someone would tell me how I can get a startup to notice me."
Make Products.
"I guess it's impossible and I'll never break into the industry."
MAKE PRODUCTS.
Courtesy of @edbrisson's wonderful thread on breaking into comics – https://t.co/TgNblNSCBj – here is why the same applies to Product Management, too.
"I really want to break into comics"
— Ed Brisson (@edbrisson) December 4, 2018
make comics.
"If only someone would tell me how I can get an editor to notice me."
Make Comics.
"I guess it's impossible and I'll never break into the industry."
MAKE COMICS.
There is no better way of learning the craft of product, or proving your potential to employers, than just doing it.
You do not need anybody's permission. We don't have diplomas, nor doctorates. We can barely agree on a single standard of what a Product Manager is supposed to do.
But – there is at least one blindingly obvious industry consensus – a Product Manager makes Products.
And they don't need to be kept at the exact right temperature, given endless resource, or carefully protected in order to do this.
They find their own way.


