
Did we get dietary saturated fats all wrong? The #HADLmodel provides a new understanding and an opportunity to get it right. THREAD👇👇👇
@simondankel @kariannesve
More from Science

Localized Surface Plasmon Resonance - an overview | ScienceDirect Topics
https://t.co/mzS7vVSREJ
https://t.co/353PdAX2fa
https://t.co/3yBImjOdd4
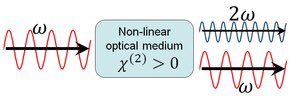
In some cases, almost 100% of the light energy can be converted to the second harmonic frequency. These cases typically involve intense pulsed laser beams passing through large crystals, and careful alignment to obtain phase matching.
https://t.co/mzS7vVSREJ
https://t.co/353PdAX2fa
https://t.co/3yBImjOdd4
In some cases, almost 100% of the light energy can be converted to the second harmonic frequency. These cases typically involve intense pulsed laser beams passing through large crystals, and careful alignment to obtain phase matching.

https://t.co/hXlo8qgkD0
Look like that they got a classical case of PCR Cross-Contamination.
They had 2 fabricated samples (SRX9714436 and SRX9714921) on the same PCR run. Alongside with Lung07. They did not perform metagenomic sequencing on the “feces” and they did not get

A positive oral or anal swab from anywhere in their sampling. Feces came from anus and if these were positive the anal swabs must also be positive. Clearly it got there after the NA have been extracted and were from the very low-level degraded RNA which were mutagenized from
The Taq. https://t.co/yKXCgiT29w to see SRX9714921 and SRX9714436.
Human+Mouse in the positive SRA, human in both of them. Seeing human+mouse in identical proportions across 3 different sequencers (PRJNA573298, A22, SEX9714436) are pretty straight indication that the originals
Were already contaminated with Human and mouse from the very beginning, and that this contamination is due to dishonesty in the sample handling process which prescribe a spiking of samples in ACE2-HEK293T/A549, VERO E6 and Human lung xenograft mouse.
The “lineages” they claimed to have found aren’t mutational lineages at all—all the mutations they see on these sequences were unique to that specific sequence, and are the result of RNA degradation and from the Taq polymerase errors accumulated from the nested PCR process

Look like that they got a classical case of PCR Cross-Contamination.
They had 2 fabricated samples (SRX9714436 and SRX9714921) on the same PCR run. Alongside with Lung07. They did not perform metagenomic sequencing on the “feces” and they did not get
A positive oral or anal swab from anywhere in their sampling. Feces came from anus and if these were positive the anal swabs must also be positive. Clearly it got there after the NA have been extracted and were from the very low-level degraded RNA which were mutagenized from
The Taq. https://t.co/yKXCgiT29w to see SRX9714921 and SRX9714436.
Human+Mouse in the positive SRA, human in both of them. Seeing human+mouse in identical proportions across 3 different sequencers (PRJNA573298, A22, SEX9714436) are pretty straight indication that the originals
Were already contaminated with Human and mouse from the very beginning, and that this contamination is due to dishonesty in the sample handling process which prescribe a spiking of samples in ACE2-HEK293T/A549, VERO E6 and Human lung xenograft mouse.
The “lineages” they claimed to have found aren’t mutational lineages at all—all the mutations they see on these sequences were unique to that specific sequence, and are the result of RNA degradation and from the Taq polymerase errors accumulated from the nested PCR process

"NO LONGER BEST IN THE WORLD"
UNEP's new Human Development Index includes a new (separate) index: Planetary pressures-adjusted HDI (PHDI). News in Norway is that its position drops from #1 to #16 because of this, while Ireland rises from #2 to #1.
Why?
https://t.co/aVraIEzRfh

Check out Norway's 'Domestic Material Consumption'. Fossil fuels are no different here to Ireland's. What's different is this huge 'non-metallic minerals' category.
(Note also the jump in 1998, suggesting data problems.)
https://t.co/5QvzONbqmN
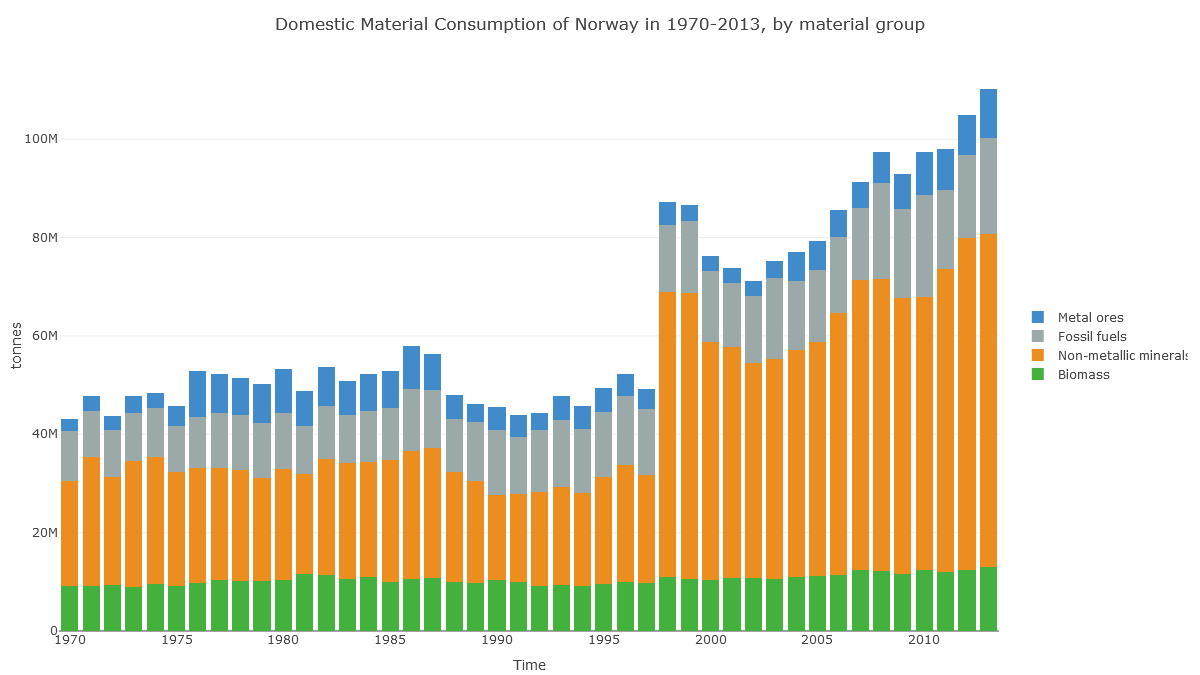
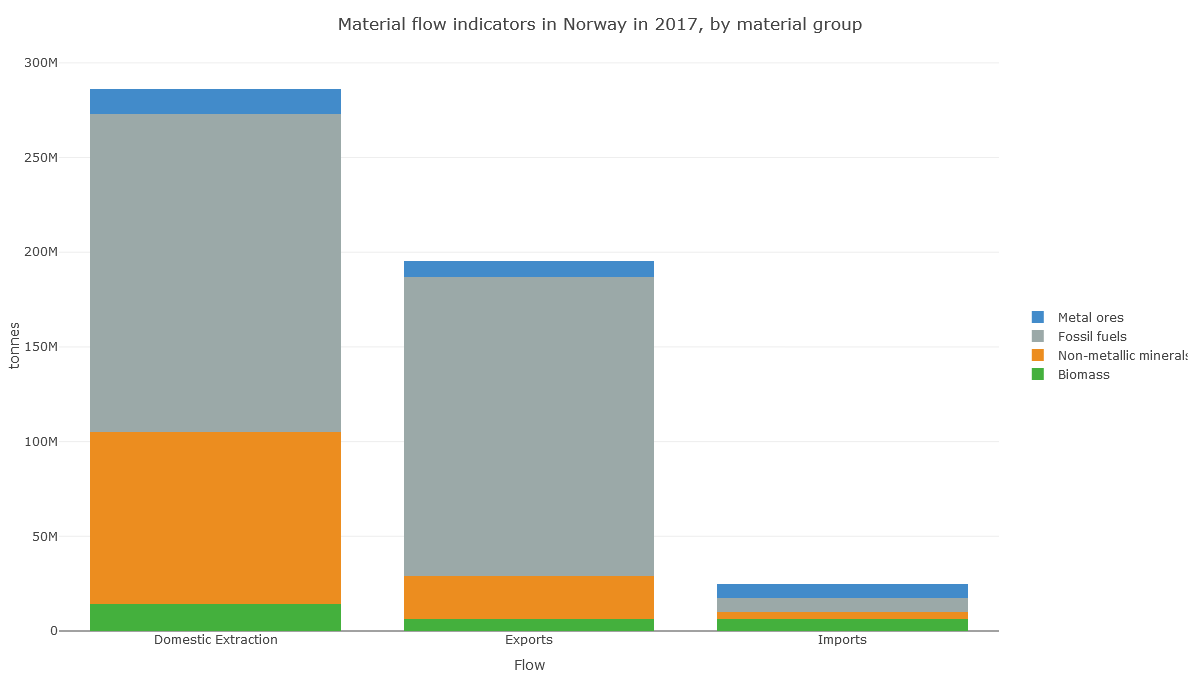
In Norway's case, it looks like the apparent consumption equation (production+imports-exports) for non-metal minerals is dominated by production: extraction of material in Norway.
https://t.co/5QvzONbqmN
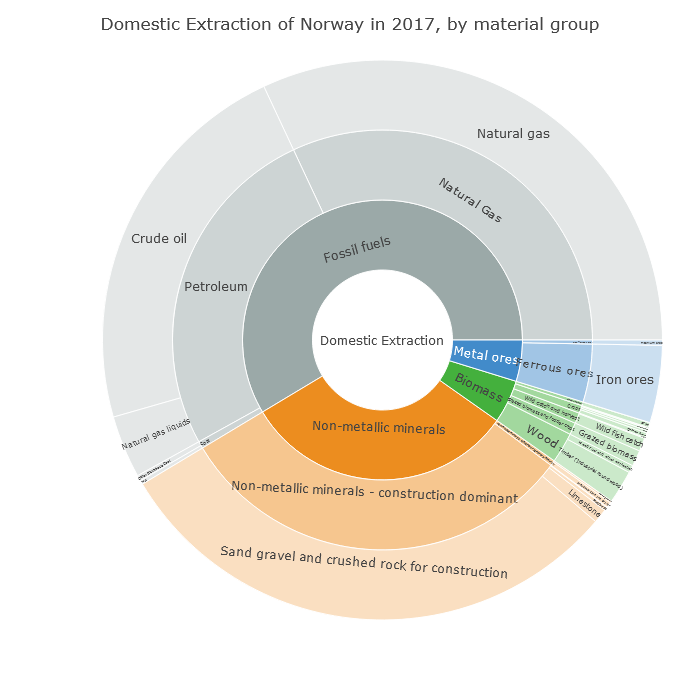
And here we see that this production of non-metallic minerals is sand, gravel and crushed rock for construction. So it's about Norway's geology.
https://t.co/y6rqWmFVWc
Norway drops 15 places on the PHDI list not because of its CO₂ emissions (fairly high at 41st highest in the world per capita), but because of its geology, because it shifts a lot of rock whenever it builds anything.
UNEP's new Human Development Index includes a new (separate) index: Planetary pressures-adjusted HDI (PHDI). News in Norway is that its position drops from #1 to #16 because of this, while Ireland rises from #2 to #1.
Why?
https://t.co/aVraIEzRfh
Check out Norway's 'Domestic Material Consumption'. Fossil fuels are no different here to Ireland's. What's different is this huge 'non-metallic minerals' category.
(Note also the jump in 1998, suggesting data problems.)
https://t.co/5QvzONbqmN
In Norway's case, it looks like the apparent consumption equation (production+imports-exports) for non-metal minerals is dominated by production: extraction of material in Norway.
https://t.co/5QvzONbqmN
And here we see that this production of non-metallic minerals is sand, gravel and crushed rock for construction. So it's about Norway's geology.
https://t.co/y6rqWmFVWc
Norway drops 15 places on the PHDI list not because of its CO₂ emissions (fairly high at 41st highest in the world per capita), but because of its geology, because it shifts a lot of rock whenever it builds anything.



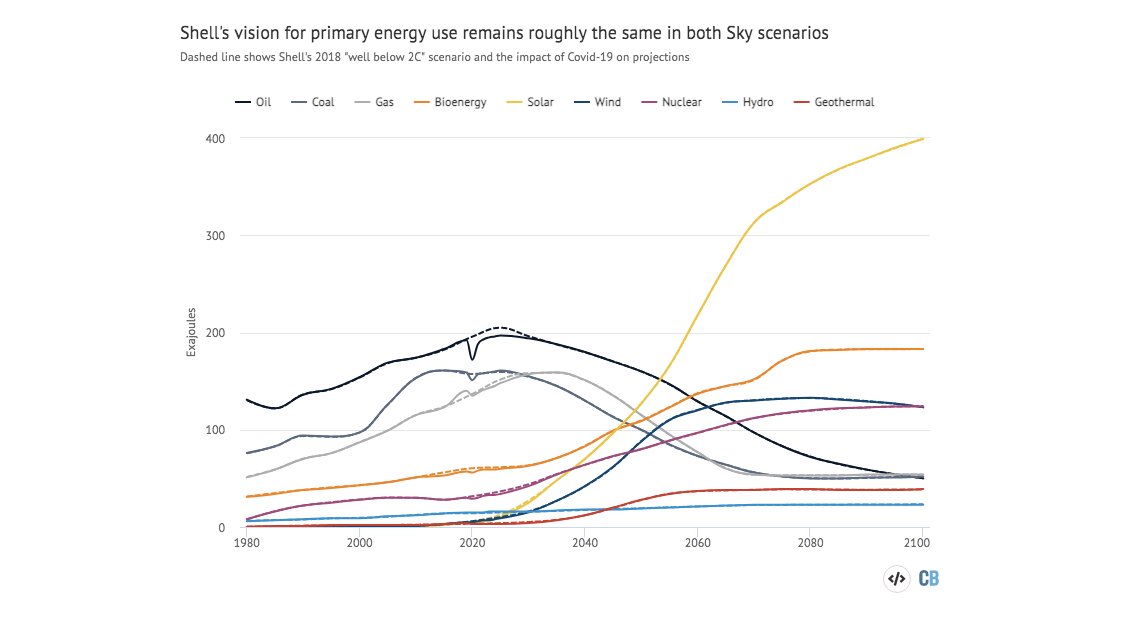
I took a look at Shell's first ever 1.5C scenario and found that it is... remarkably similar to its “well-below 2C” scenario.
Oil, gas, coal, solar.... all basically unchanged.
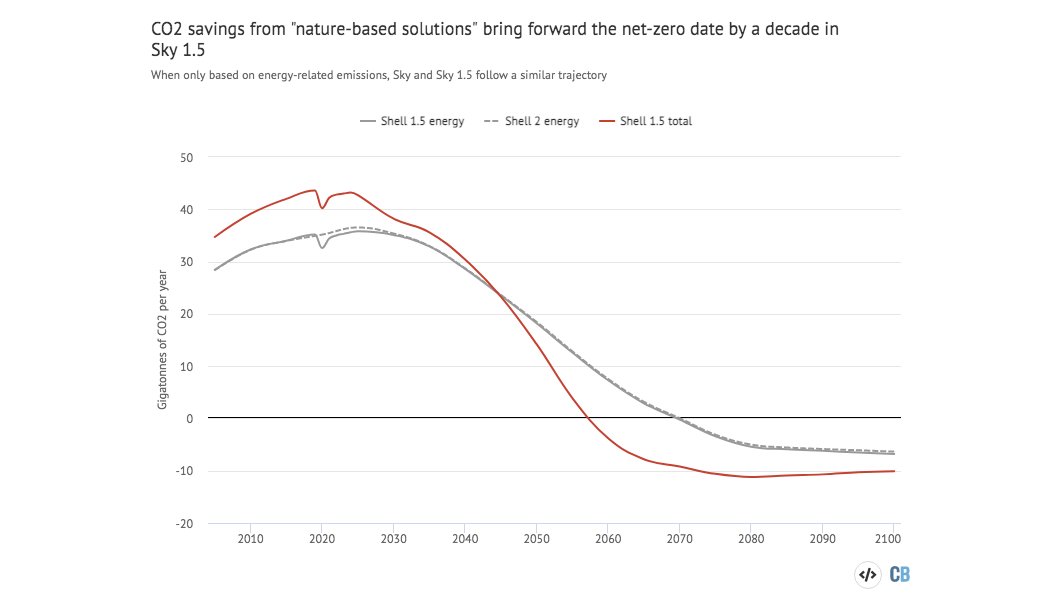
The key difference: A new forest the size of Brazil to suck up the extra CO2.
Including "nature-based solutions" in the outlook brings forward the date for net-zero emissions to 2058.
Without them their pathway for CO2 emissions is the same as the previous one.
(It's also towards the higher end of 1.5C emissions pathways.)
The "Brazil-sized" forest idea isn't actually new, it has been kicking around for a couple of years.
It was referenced in the "well-below 2C" scenario although not formally included in it, and Shell's CEO has been framing it as the only viable way of getting to 1.5C.

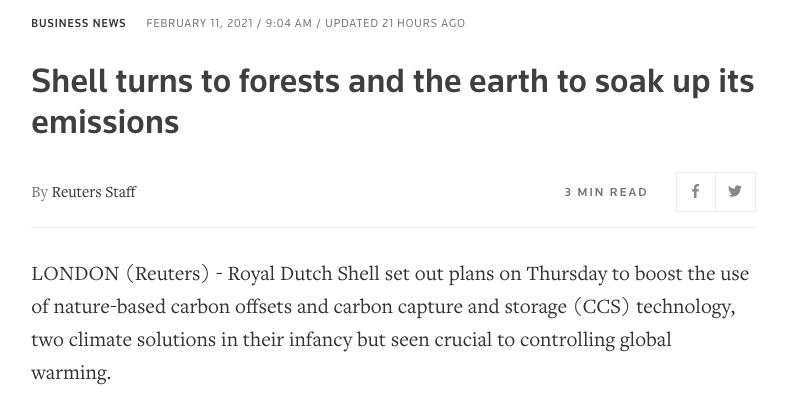
Fine, but who is going to plant all those trees? Well... Shell says it will plant some of them.
Only yesterday Shell said forests were a key part of its net-zero strategy.
Not everyone is convinced though
https://t.co/RaJm7tOHxb
Given that Shell's 1.5C scenario also sees a big scaling up of bioenergy, the question remains: where are all those trees and bioenergy crops going to go?

Oil, gas, coal, solar.... all basically unchanged.
The key difference: A new forest the size of Brazil to suck up the extra CO2.
Including "nature-based solutions" in the outlook brings forward the date for net-zero emissions to 2058.
Without them their pathway for CO2 emissions is the same as the previous one.
(It's also towards the higher end of 1.5C emissions pathways.)
The "Brazil-sized" forest idea isn't actually new, it has been kicking around for a couple of years.
It was referenced in the "well-below 2C" scenario although not formally included in it, and Shell's CEO has been framing it as the only viable way of getting to 1.5C.
Fine, but who is going to plant all those trees? Well... Shell says it will plant some of them.
Only yesterday Shell said forests were a key part of its net-zero strategy.
Not everyone is convinced though
https://t.co/RaJm7tOHxb
Shell plans to use forests to remove 120 Mt/yr of CO2 by 2030.
— Greg Muttitt (@FuelOnTheFire) February 12, 2021
Appropriate land for forestation is finite, and risks competition with food production and human rights of current land owners/users, esp Indigenous
Given that Shell's 1.5C scenario also sees a big scaling up of bioenergy, the question remains: where are all those trees and bioenergy crops going to go?
You May Also Like






Fake chats claiming to be from the Irish African community are being disseminated by the far right in order to suggest that violence is imminent from #BLM supporters. This is straight out of the QAnon and Proud Boys playbook. Spread the word. Protest safely. #georgenkencho

There is co-ordination across the far right in Ireland now to stir both left and right in the hopes of creating a race war. Think critically! Fascists see the tragic killing of #georgenkencho, the grief of his community and pending investigation as a flashpoint for action.
Across Telegram, Twitter and Facebook disinformation is being peddled on the back of these tragic events. From false photographs to the tactics ofwhite supremacy, the far right is clumsily trying to drive hate against minority groups and figureheads.
Declan Ganley’s Burkean group and the incel wing of National Party (Gearóid Murphy, Mick O’Keeffe & Co.) as well as all the usuals are concerted in their efforts to demonstrate their white supremacist cred. The quiet parts are today being said out loud.
The best thing you can do is challenge disinformation and report posts where engagement isn’t appropriate. Many of these are blatantly racist posts designed to drive recruitment to NP and other Nationalist groups. By all means protest but stay safe.
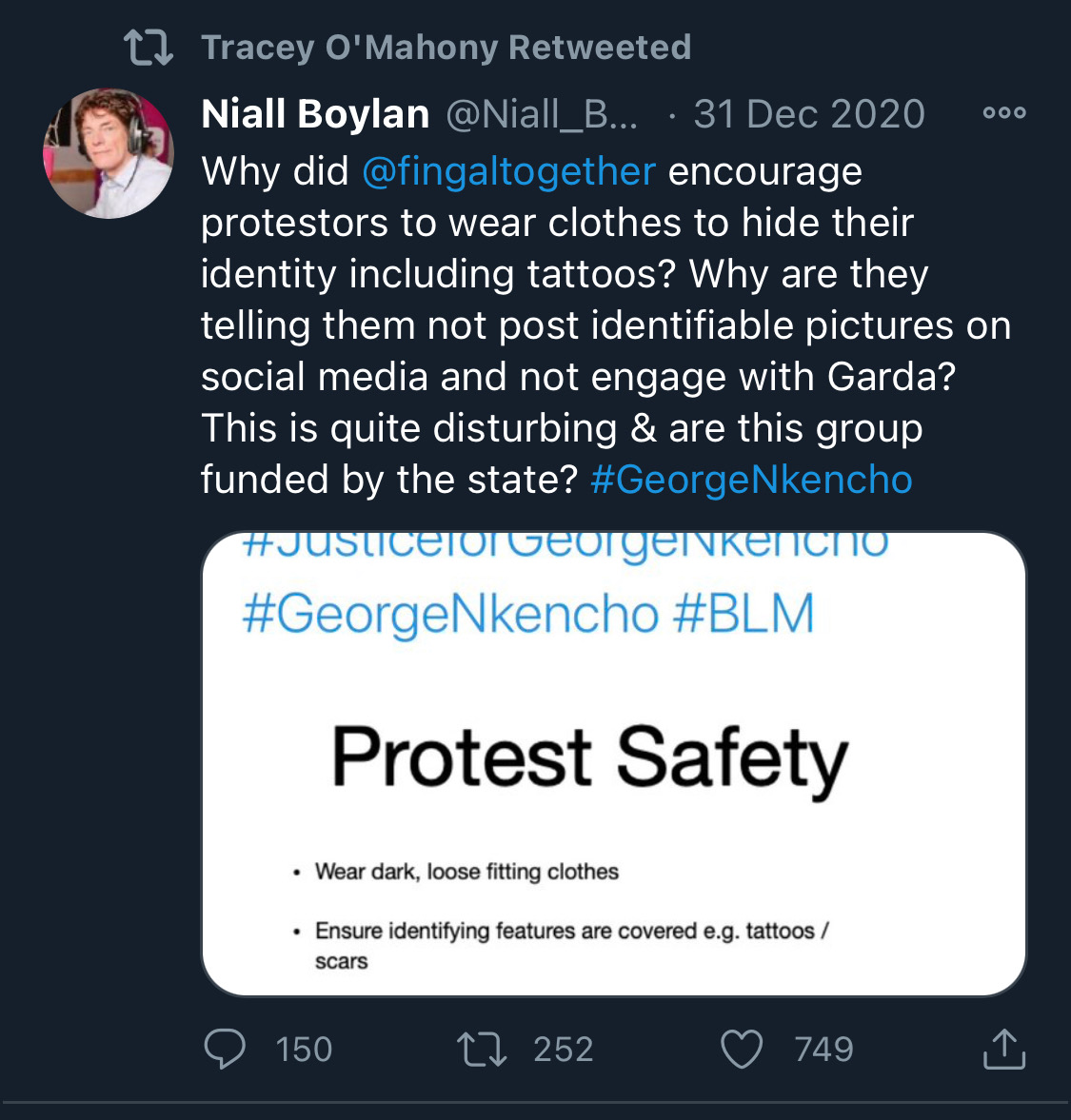
There is co-ordination across the far right in Ireland now to stir both left and right in the hopes of creating a race war. Think critically! Fascists see the tragic killing of #georgenkencho, the grief of his community and pending investigation as a flashpoint for action.
Across Telegram, Twitter and Facebook disinformation is being peddled on the back of these tragic events. From false photographs to the tactics ofwhite supremacy, the far right is clumsily trying to drive hate against minority groups and figureheads.
Be aware, the images the #farright are sharing in the hopes of starting a race war, are not of the SPAR employee that was punched. They\u2019re older photos of a Everton fan. Be aware of the information you\u2019re sharing and that it may be false. Always #factcheck #GeorgeNkencho pic.twitter.com/4c9w4CMk5h
— antifa.drone (@antifa_drone) December 31, 2020
Declan Ganley’s Burkean group and the incel wing of National Party (Gearóid Murphy, Mick O’Keeffe & Co.) as well as all the usuals are concerted in their efforts to demonstrate their white supremacist cred. The quiet parts are today being said out loud.
There is a concerted effort in far-right Telegram groups to try and incite violence on street by targetting people for racist online abuse following the killing of George Nkencho
— Mark Malone (@soundmigration) January 1, 2021
This follows on and is part of a misinformation campaign to polarise communities at this time.
The best thing you can do is challenge disinformation and report posts where engagement isn’t appropriate. Many of these are blatantly racist posts designed to drive recruitment to NP and other Nationalist groups. By all means protest but stay safe.


