
Here's more on the new mitigation scenario for 1.5C. How does it work? What would society look like? Are we willing to do what's required to stop climate breakdown? See thread.
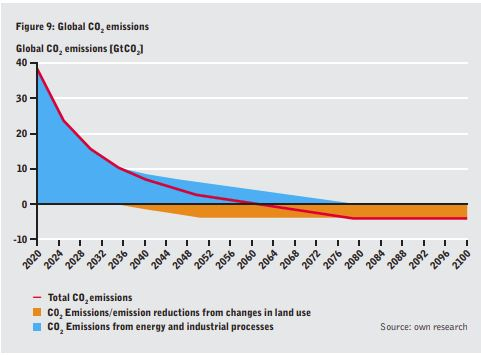
•a shift from private cars to public transportation
•reduction in flights
•smaller average house size
Meanwhile, consumption in the global South (non-Annex 1 countries) rises to converge.
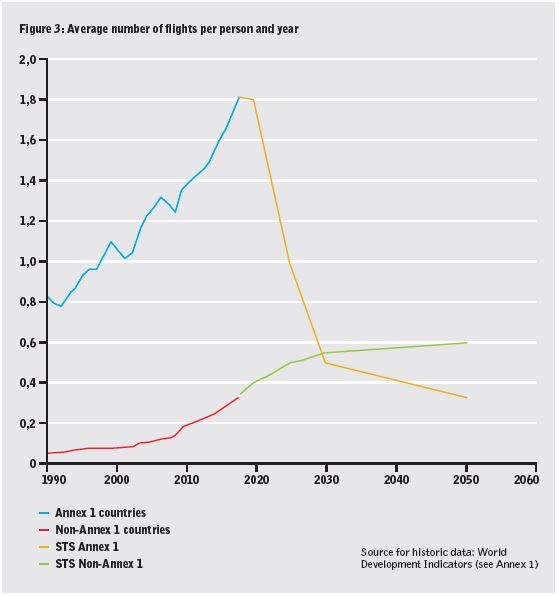
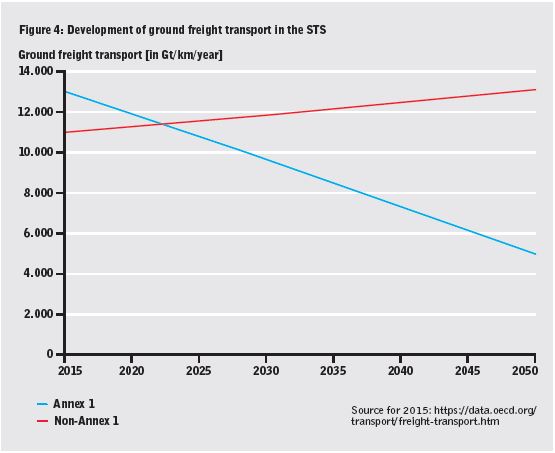
•Meat consumption in high-income nations falls by about two-thirds (with specific focus on beef).
•Significant reduction in food waste.
•Global transition from industrial farming to regenerative agricultural methods to restore soils and biodiversity.
•Universal public services
•Shorter working hours
•Basic income and maximum wage
•Radical reduction in inequality
These measures ensure that all people have access to the resources they need to live flourishing lives even as aggregate economic output declines.
More from Science

"NO LONGER BEST IN THE WORLD"
UNEP's new Human Development Index includes a new (separate) index: Planetary pressures-adjusted HDI (PHDI). News in Norway is that its position drops from #1 to #16 because of this, while Ireland rises from #2 to #1.
Why?
https://t.co/aVraIEzRfh

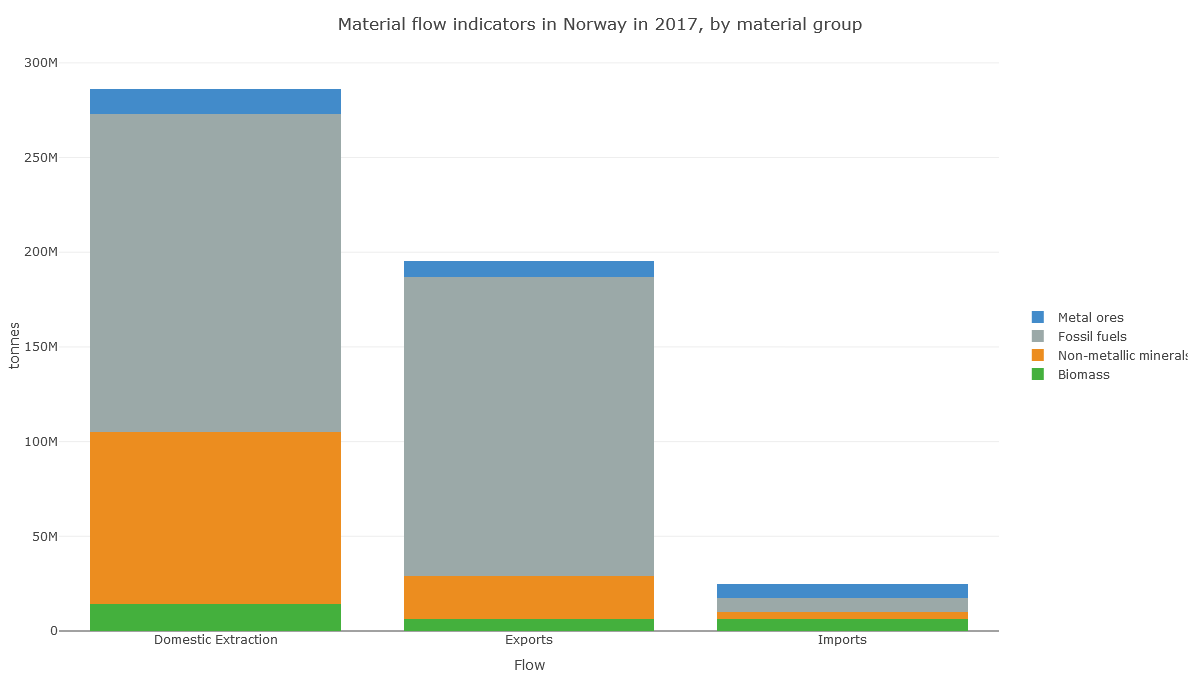
Check out Norway's 'Domestic Material Consumption'. Fossil fuels are no different here to Ireland's. What's different is this huge 'non-metallic minerals' category.
(Note also the jump in 1998, suggesting data problems.)
https://t.co/5QvzONbqmN
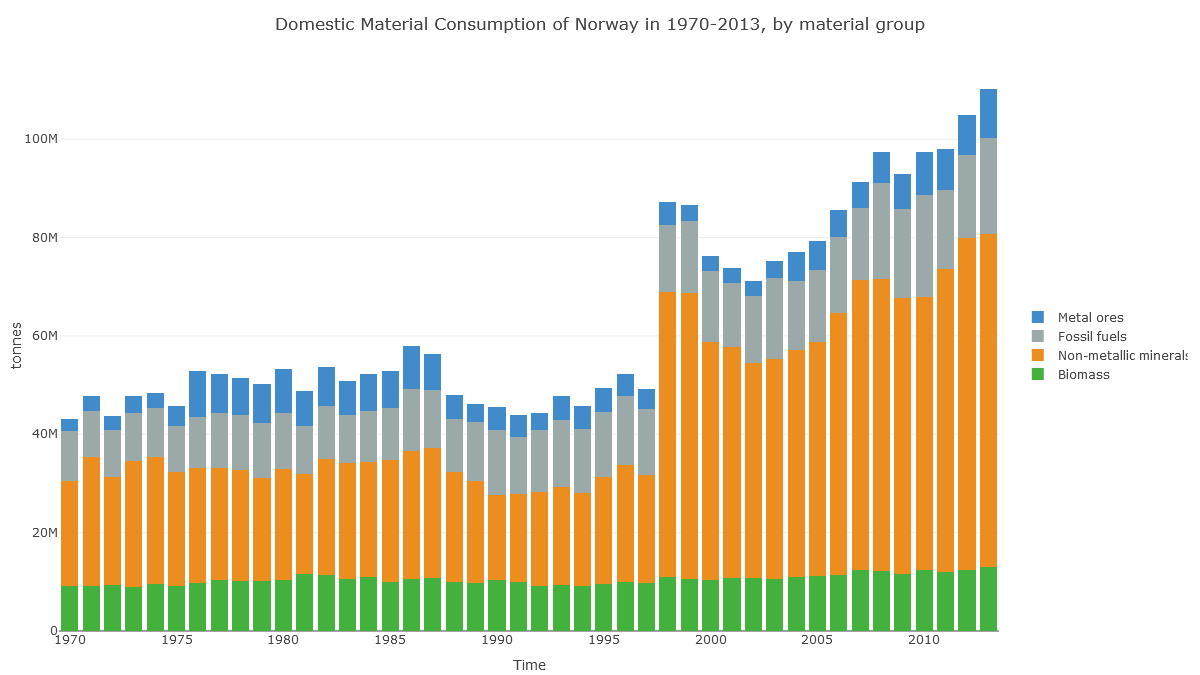
In Norway's case, it looks like the apparent consumption equation (production+imports-exports) for non-metal minerals is dominated by production: extraction of material in Norway.
https://t.co/5QvzONbqmN
And here we see that this production of non-metallic minerals is sand, gravel and crushed rock for construction. So it's about Norway's geology.
https://t.co/y6rqWmFVWc
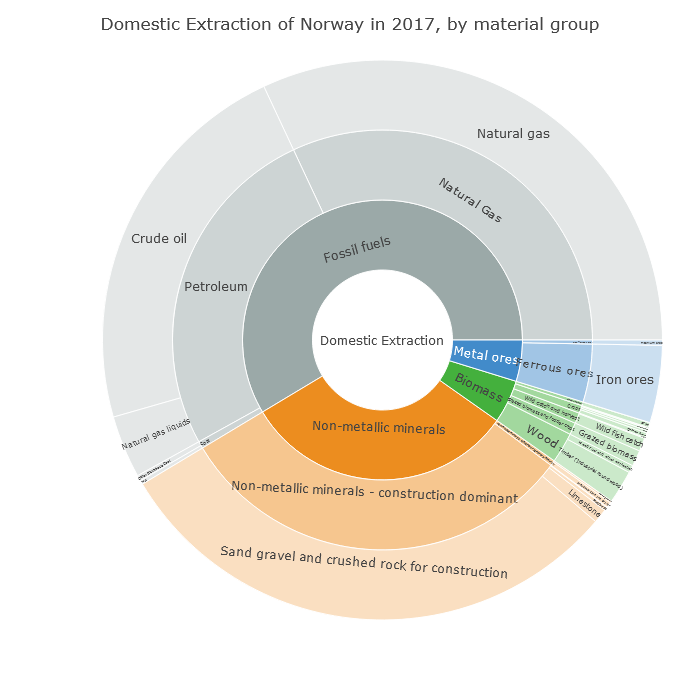
Norway drops 15 places on the PHDI list not because of its CO₂ emissions (fairly high at 41st highest in the world per capita), but because of its geology, because it shifts a lot of rock whenever it builds anything.
UNEP's new Human Development Index includes a new (separate) index: Planetary pressures-adjusted HDI (PHDI). News in Norway is that its position drops from #1 to #16 because of this, while Ireland rises from #2 to #1.
Why?
https://t.co/aVraIEzRfh
Check out Norway's 'Domestic Material Consumption'. Fossil fuels are no different here to Ireland's. What's different is this huge 'non-metallic minerals' category.
(Note also the jump in 1998, suggesting data problems.)
https://t.co/5QvzONbqmN
In Norway's case, it looks like the apparent consumption equation (production+imports-exports) for non-metal minerals is dominated by production: extraction of material in Norway.
https://t.co/5QvzONbqmN
And here we see that this production of non-metallic minerals is sand, gravel and crushed rock for construction. So it's about Norway's geology.
https://t.co/y6rqWmFVWc
Norway drops 15 places on the PHDI list not because of its CO₂ emissions (fairly high at 41st highest in the world per capita), but because of its geology, because it shifts a lot of rock whenever it builds anything.

JUST ONE PERSON—UK 🇬🇧 scientists think one immunocompromised person who cleared virus slowly & only partially wiped out an infection, leaving behind genetically-hardier viruses that rebound & learn how to survive better. That’s likely how #B117 started. 🧵 https://t.co/bMMjM8Hiuz
2) The leading hypothesis is that the new variant evolved within just one person, chronically infected with the virus for so long it was able to evolve into a new, more infectious form.
same thing happened in Boston in another immunocompromised person that was sick for 155 days.
3) What happened in Boston with one 45 year old man who was highly infectious for 155 days straight before he died... is exactly what scientists think happened in Kent, England that gave rise to #B117.
4) Doctors were shocked to find virus has evolved many different forms inside of this one immunocompromised man. 20 new mutations in one virus, akin to the #B117. This is possibly how #B1351 in South Africa 🇿🇦 and #P1 in Brazil 🇧🇷 also evolved.
5) “On its own, the appearance of a new variant in genomic databases doesn’t tell us much. “That’s just one genome amongst thousands every week. It wouldn’t necessarily stick out,” says Oliver Pybus, a professor of evolution and infectious disease at Oxford.
2) The leading hypothesis is that the new variant evolved within just one person, chronically infected with the virus for so long it was able to evolve into a new, more infectious form.
same thing happened in Boston in another immunocompromised person that was sick for 155 days.
3) What happened in Boston with one 45 year old man who was highly infectious for 155 days straight before he died... is exactly what scientists think happened in Kent, England that gave rise to #B117.
Immunocompromised 45 year old suffered from #COVID19 for 155 days before he died. The virus was changing very quickly inside the man's body\u2014it acquired a big cluster of >20 mutations\u2014resembled the same ones seen in #B117 & #B1351. (NPR audio Part 1 of 2)\U0001f9f5https://t.co/7kWiBZ1xGk pic.twitter.com/ZJ7AExB78Y
— Eric Feigl-Ding (@DrEricDing) February 8, 2021
4) Doctors were shocked to find virus has evolved many different forms inside of this one immunocompromised man. 20 new mutations in one virus, akin to the #B117. This is possibly how #B1351 in South Africa 🇿🇦 and #P1 in Brazil 🇧🇷 also evolved.
2) NPR report audio part 2 of 2:
— Eric Feigl-Ding (@DrEricDing) February 8, 2021
Dr. Li couldn't believe what they found. "I was shocked," he says. "When I saw the virus sequences, I knew that we were dealing with something completely different and potentially very important." pic.twitter.com/HT3Yt6djFd
5) “On its own, the appearance of a new variant in genomic databases doesn’t tell us much. “That’s just one genome amongst thousands every week. It wouldn’t necessarily stick out,” says Oliver Pybus, a professor of evolution and infectious disease at Oxford.

An interesting thing about carp is that they can go into anoxic hibernation and switch to an anaerobic metabolism based on converting glycogen to ethanol.
The waste ethanol is diffused out the gills
https://t.co/V3D1umHf04
Carp can switch over to an anaerobic metabolism and quietly exhale booze until the situation gets better.
They basically evolved the same metabolic pathway as yeast, independently.
In theory, if you spent a few thousand years breeding carp for it, you could use them to make booze.
They'd be enormous, almost entirely glycogen deposits with a fish added as an afterthought.
The really interesting thing about anaerobic carp, is that they can go 4-5 months without oxygen by relying on liver glycogen.
You, a human, have only about 100 grams of glycogen in your liver, about 400 more grams in your skeletal muscles. Call it 500 grams total.
In humans, glycogen is also burned for energy. This is where the marathon runner's bonk comes from: you only have about 2,000 calories worth, and running a marathon burns those 2,000 calories.
The waste ethanol is diffused out the gills
https://t.co/V3D1umHf04
Carp can switch over to an anaerobic metabolism and quietly exhale booze until the situation gets better.
They basically evolved the same metabolic pathway as yeast, independently.
In theory, if you spent a few thousand years breeding carp for it, you could use them to make booze.
They'd be enormous, almost entirely glycogen deposits with a fish added as an afterthought.
The really interesting thing about anaerobic carp, is that they can go 4-5 months without oxygen by relying on liver glycogen.
You, a human, have only about 100 grams of glycogen in your liver, about 400 more grams in your skeletal muscles. Call it 500 grams total.
In humans, glycogen is also burned for energy. This is where the marathon runner's bonk comes from: you only have about 2,000 calories worth, and running a marathon burns those 2,000 calories.



You May Also Like

Neo-nazi group #PatriotFront held a photo op in #Chicago last weekend & is currently marching around #DC so it's as good time as any to compile a list of their identified members for folks to watch for
Who are these chuds?
Patriot Front broke away from white nationalist org Vanguard America following #unitetheright in #charlottesville after James Alex Fields was seen with a VA shield before driving his car into a crowd, murdering Heather Heyer & injuring dozens of others
Syed Robbie Javid a.k.a. Sayed Robbie Javid or Robbie Javid of Alexandria,
Antoine Bernard Renard (a.k.a. “Charlemagne MD” on Discord) from Rockville, MD.
https://t.co/ykEjdZFDi6

Brandon Troy Higgs, 25, from Reisterstown,
Who are these chuds?
Patriot Front broke away from white nationalist org Vanguard America following #unitetheright in #charlottesville after James Alex Fields was seen with a VA shield before driving his car into a crowd, murdering Heather Heyer & injuring dozens of others
Syed Robbie Javid a.k.a. Sayed Robbie Javid or Robbie Javid of Alexandria,
Happy Monday everyone :-) Let's ring in September by reacquainting ourselves with Virginia neo-Nazi and NSC Dixie affiliate Sayed "Robbie" Javid, now known by "Reform the States". Robbie is an explicitly genocidal neo-Nazi, so lets get to know him a bit better!
— Garfield but Anti-Fascist (@AntifaGarfield) August 31, 2020
CW on this thread pic.twitter.com/3gzxrIo9HD
Antoine Bernard Renard (a.k.a. “Charlemagne MD” on Discord) from Rockville, MD.
https://t.co/ykEjdZFDi6
Brandon Troy Higgs, 25, from Reisterstown,












I hate when I learn something new (to me) & stunning about the Jeff Epstein network (h/t MoodyKnowsNada.)
Where to begin?
So our new Secretary of State Anthony Blinken's stepfather, Samuel Pisar, was "longtime lawyer and confidant of...Robert Maxwell," Ghislaine Maxwell's Dad.

"Pisar was one of the last people to speak to Maxwell, by phone, probably an hour before the chairman of Mirror Group Newspapers fell off his luxury yacht the Lady Ghislaine on 5 November, 1991." https://t.co/DAEgchNyTP

OK, so that's just a coincidence. Moving on, Anthony Blinken "attended the prestigious Dalton School in New York City"...wait, what? https://t.co/DnE6AvHmJg
Dalton School...Dalton School...rings a
Oh that's right.
The dad of the U.S. Attorney General under both George W. Bush & Donald Trump, William Barr, was headmaster of the Dalton School.
Donald Barr was also quite a
I'm not going to even mention that Blinken's stepdad Sam Pisar's name was in Epstein's "black book."
Lots of names in that book. I mean, for example, Cuomo, Trump, Clinton, Prince Andrew, Bill Cosby, Woody Allen - all in that book, and their reputations are spotless.

Where to begin?
So our new Secretary of State Anthony Blinken's stepfather, Samuel Pisar, was "longtime lawyer and confidant of...Robert Maxwell," Ghislaine Maxwell's Dad.
"Pisar was one of the last people to speak to Maxwell, by phone, probably an hour before the chairman of Mirror Group Newspapers fell off his luxury yacht the Lady Ghislaine on 5 November, 1991." https://t.co/DAEgchNyTP
OK, so that's just a coincidence. Moving on, Anthony Blinken "attended the prestigious Dalton School in New York City"...wait, what? https://t.co/DnE6AvHmJg
Dalton School...Dalton School...rings a
Oh that's right.
The dad of the U.S. Attorney General under both George W. Bush & Donald Trump, William Barr, was headmaster of the Dalton School.
Donald Barr was also quite a
Donald Barr had a way with words. pic.twitter.com/JdRBwXPhJn
— Rudy Havenstein, listening to Nas all day. (@RudyHavenstein) September 17, 2020
I'm not going to even mention that Blinken's stepdad Sam Pisar's name was in Epstein's "black book."
Lots of names in that book. I mean, for example, Cuomo, Trump, Clinton, Prince Andrew, Bill Cosby, Woody Allen - all in that book, and their reputations are spotless.




