
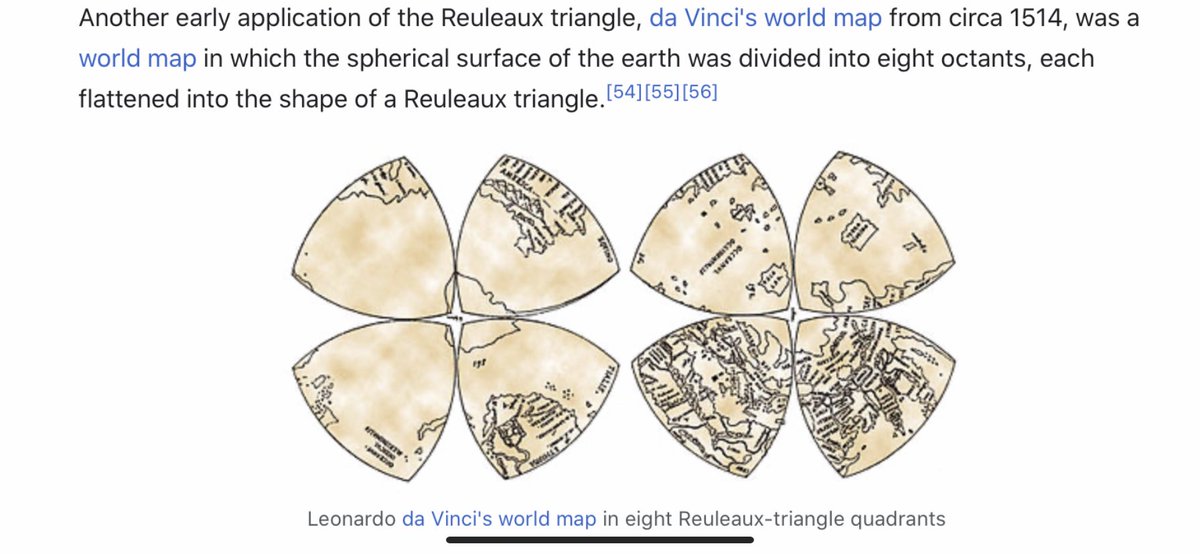
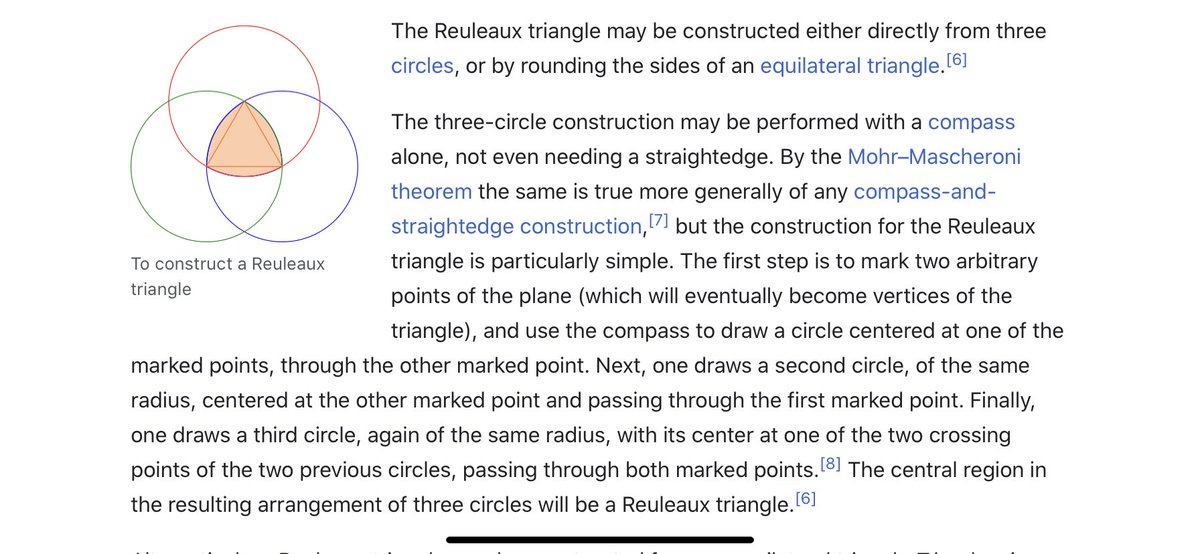
I think we have to expand our thinking about the toroidal sphere even more. When looking at maps, I noticed the da Vinci map, from 1514, which uses the Reuleaux Triangle. This triangle is formed from 3 intersecting circles, and is in the center of a trefoil.




More from Science

Hugh Everett's birthday! Pioneer of the Many-Worlds Interpretation of quantum mechanics. Let us celebrate by thinking about ontological extravagance. I will do so by way of analogy, because I have found that everyone loves analogies and nobody ever willfully misconstrues them.
We look at the night sky and see photons arriving to us, emitted by distant stars. Let's contrast two different theories about how stars emit photons.
One theory says, we know how stars shine, and our equations predict that they emit photons roughly uniformly in all directions. Call this the "Many-Photons Interpretation" (MPI).
But! Others object. That is *so many photons*. Most of which we don't observe, and can't observe, since they're moving away at the speed of light. It's too ontologically extravagant to posit a huge number of unobservable things!
So they suggest a "Photon Collapse Interpretation." According to this theory, the photons emitted toward us actually exist. But photons that would be emitted in directions we will never observe simply collapse into utter non-existence.
The physicist Hugh Everett III was born #OTD in 1930. His \u201crelative state\u201d formulation of quantum mechanics, which we now call the \u201cMany Worlds Interpretation,\u201d was published in 1957. pic.twitter.com/ZqMsZcPJDG
— Robert McNees, the bastegod (@mcnees) November 11, 2020
We look at the night sky and see photons arriving to us, emitted by distant stars. Let's contrast two different theories about how stars emit photons.
One theory says, we know how stars shine, and our equations predict that they emit photons roughly uniformly in all directions. Call this the "Many-Photons Interpretation" (MPI).
But! Others object. That is *so many photons*. Most of which we don't observe, and can't observe, since they're moving away at the speed of light. It's too ontologically extravagant to posit a huge number of unobservable things!
So they suggest a "Photon Collapse Interpretation." According to this theory, the photons emitted toward us actually exist. But photons that would be emitted in directions we will never observe simply collapse into utter non-existence.

https://t.co/hXlo8qgkD0
Look like that they got a classical case of PCR Cross-Contamination.
They had 2 fabricated samples (SRX9714436 and SRX9714921) on the same PCR run. Alongside with Lung07. They did not perform metagenomic sequencing on the “feces” and they did not get

A positive oral or anal swab from anywhere in their sampling. Feces came from anus and if these were positive the anal swabs must also be positive. Clearly it got there after the NA have been extracted and were from the very low-level degraded RNA which were mutagenized from
The Taq. https://t.co/yKXCgiT29w to see SRX9714921 and SRX9714436.
Human+Mouse in the positive SRA, human in both of them. Seeing human+mouse in identical proportions across 3 different sequencers (PRJNA573298, A22, SEX9714436) are pretty straight indication that the originals
Were already contaminated with Human and mouse from the very beginning, and that this contamination is due to dishonesty in the sample handling process which prescribe a spiking of samples in ACE2-HEK293T/A549, VERO E6 and Human lung xenograft mouse.
The “lineages” they claimed to have found aren’t mutational lineages at all—all the mutations they see on these sequences were unique to that specific sequence, and are the result of RNA degradation and from the Taq polymerase errors accumulated from the nested PCR process

Look like that they got a classical case of PCR Cross-Contamination.
They had 2 fabricated samples (SRX9714436 and SRX9714921) on the same PCR run. Alongside with Lung07. They did not perform metagenomic sequencing on the “feces” and they did not get
A positive oral or anal swab from anywhere in their sampling. Feces came from anus and if these were positive the anal swabs must also be positive. Clearly it got there after the NA have been extracted and were from the very low-level degraded RNA which were mutagenized from
The Taq. https://t.co/yKXCgiT29w to see SRX9714921 and SRX9714436.
Human+Mouse in the positive SRA, human in both of them. Seeing human+mouse in identical proportions across 3 different sequencers (PRJNA573298, A22, SEX9714436) are pretty straight indication that the originals
Were already contaminated with Human and mouse from the very beginning, and that this contamination is due to dishonesty in the sample handling process which prescribe a spiking of samples in ACE2-HEK293T/A549, VERO E6 and Human lung xenograft mouse.
The “lineages” they claimed to have found aren’t mutational lineages at all—all the mutations they see on these sequences were unique to that specific sequence, and are the result of RNA degradation and from the Taq polymerase errors accumulated from the nested PCR process


Hard agree. And if this is useful, let me share something that often gets omitted (not by @kakape).
Variants always emerge, & are not good or bad, but expected. The challenge is figuring out which variants are bad, and that can't be done with sequence alone.
You can't just look at a sequence and say, "Aha! A mutation in spike. This must be more transmissible or can evade antibody neutralization." Sure, we can use computational models to try and predict the functional consequence of a given mutation, but models are often wrong.
The virus acquires mutations randomly every time it replicates. Many mutations don't change the virus at all. Others may change it in a way that have no consequences for human transmission or disease. But you can't tell just looking at sequence alone.
In order to determine the functional impact of a mutation, you need to actually do experiments. You can look at some effects in cell culture, but to address questions relating to transmission or disease, you have to use animal models.
The reason people were concerned initially about B.1.1.7 is because of epidemiological evidence showing that it rapidly became dominant in one area. More rapidly that could be explained unless it had some kind of advantage that allowed it to outcompete other circulating variants.
Variants always emerge, & are not good or bad, but expected. The challenge is figuring out which variants are bad, and that can't be done with sequence alone.
Feels like the next thing we're going to need is a ranking system for how concerning "variants of concern\u201d actually are.
— Kai Kupferschmidt (@kakape) January 15, 2021
A lot of constellations of mutations are concerning, but people are lumping together variants with vastly different levels of evidence that we need to worry.
You can't just look at a sequence and say, "Aha! A mutation in spike. This must be more transmissible or can evade antibody neutralization." Sure, we can use computational models to try and predict the functional consequence of a given mutation, but models are often wrong.
The virus acquires mutations randomly every time it replicates. Many mutations don't change the virus at all. Others may change it in a way that have no consequences for human transmission or disease. But you can't tell just looking at sequence alone.
In order to determine the functional impact of a mutation, you need to actually do experiments. You can look at some effects in cell culture, but to address questions relating to transmission or disease, you have to use animal models.
The reason people were concerned initially about B.1.1.7 is because of epidemiological evidence showing that it rapidly became dominant in one area. More rapidly that could be explained unless it had some kind of advantage that allowed it to outcompete other circulating variants.

JUST ONE PERSON—UK 🇬🇧 scientists think one immunocompromised person who cleared virus slowly & only partially wiped out an infection, leaving behind genetically-hardier viruses that rebound & learn how to survive better. That’s likely how #B117 started. 🧵 https://t.co/bMMjM8Hiuz
2) The leading hypothesis is that the new variant evolved within just one person, chronically infected with the virus for so long it was able to evolve into a new, more infectious form.
same thing happened in Boston in another immunocompromised person that was sick for 155 days.
3) What happened in Boston with one 45 year old man who was highly infectious for 155 days straight before he died... is exactly what scientists think happened in Kent, England that gave rise to #B117.
4) Doctors were shocked to find virus has evolved many different forms inside of this one immunocompromised man. 20 new mutations in one virus, akin to the #B117. This is possibly how #B1351 in South Africa 🇿🇦 and #P1 in Brazil 🇧🇷 also evolved.
5) “On its own, the appearance of a new variant in genomic databases doesn’t tell us much. “That’s just one genome amongst thousands every week. It wouldn’t necessarily stick out,” says Oliver Pybus, a professor of evolution and infectious disease at Oxford.
2) The leading hypothesis is that the new variant evolved within just one person, chronically infected with the virus for so long it was able to evolve into a new, more infectious form.
same thing happened in Boston in another immunocompromised person that was sick for 155 days.
3) What happened in Boston with one 45 year old man who was highly infectious for 155 days straight before he died... is exactly what scientists think happened in Kent, England that gave rise to #B117.
Immunocompromised 45 year old suffered from #COVID19 for 155 days before he died. The virus was changing very quickly inside the man's body\u2014it acquired a big cluster of >20 mutations\u2014resembled the same ones seen in #B117 & #B1351. (NPR audio Part 1 of 2)\U0001f9f5https://t.co/7kWiBZ1xGk pic.twitter.com/ZJ7AExB78Y
— Eric Feigl-Ding (@DrEricDing) February 8, 2021
4) Doctors were shocked to find virus has evolved many different forms inside of this one immunocompromised man. 20 new mutations in one virus, akin to the #B117. This is possibly how #B1351 in South Africa 🇿🇦 and #P1 in Brazil 🇧🇷 also evolved.
2) NPR report audio part 2 of 2:
— Eric Feigl-Ding (@DrEricDing) February 8, 2021
Dr. Li couldn't believe what they found. "I was shocked," he says. "When I saw the virus sequences, I knew that we were dealing with something completely different and potentially very important." pic.twitter.com/HT3Yt6djFd
5) “On its own, the appearance of a new variant in genomic databases doesn’t tell us much. “That’s just one genome amongst thousands every week. It wouldn’t necessarily stick out,” says Oliver Pybus, a professor of evolution and infectious disease at Oxford.













