
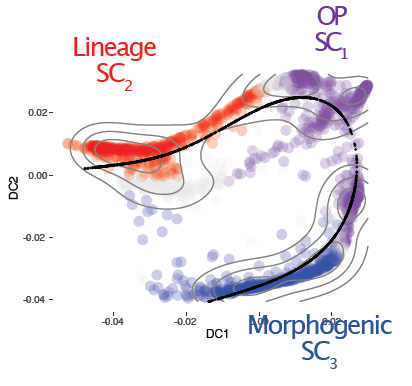
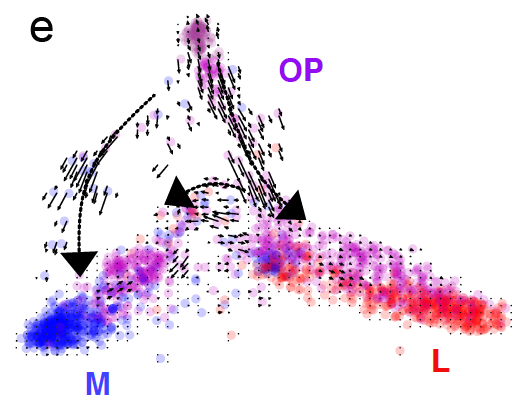
Extremely excited today to reveal the first of two great works (magnus opera?), just posted on bioarxiv, applying regulatory network analysis techniques to PDAC expression data to dissect the underlying biology of the disease. Tweetorial time! 1/






More from Science



💥and so it begins..💥
It's time, my friends 🤩🤩
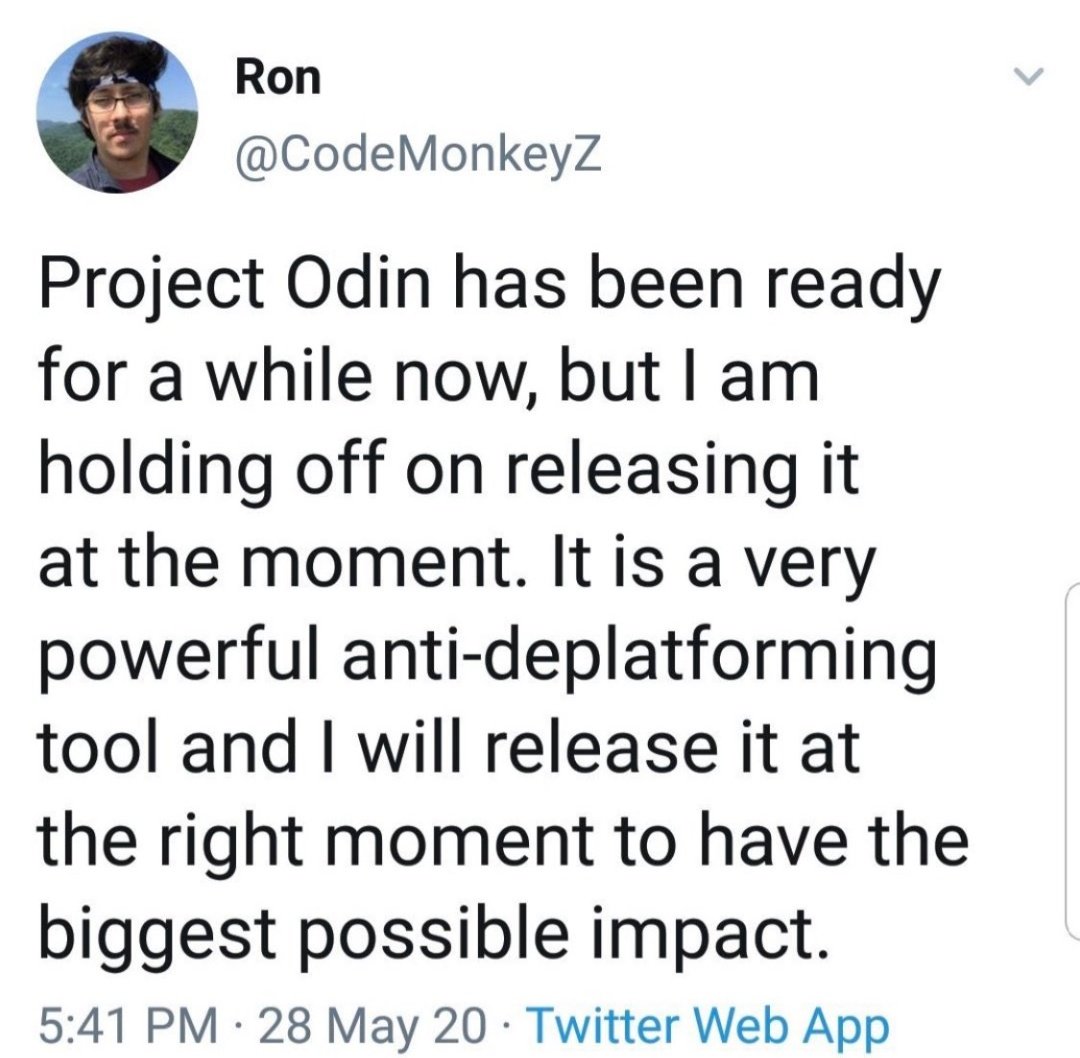
[Thread] #ProjectOdin
https://t.co/fO90N78fta
new quantum-based internet #ElonMusk #QVS #QFS
Political justification ⏬⏬
#ProjectOdin
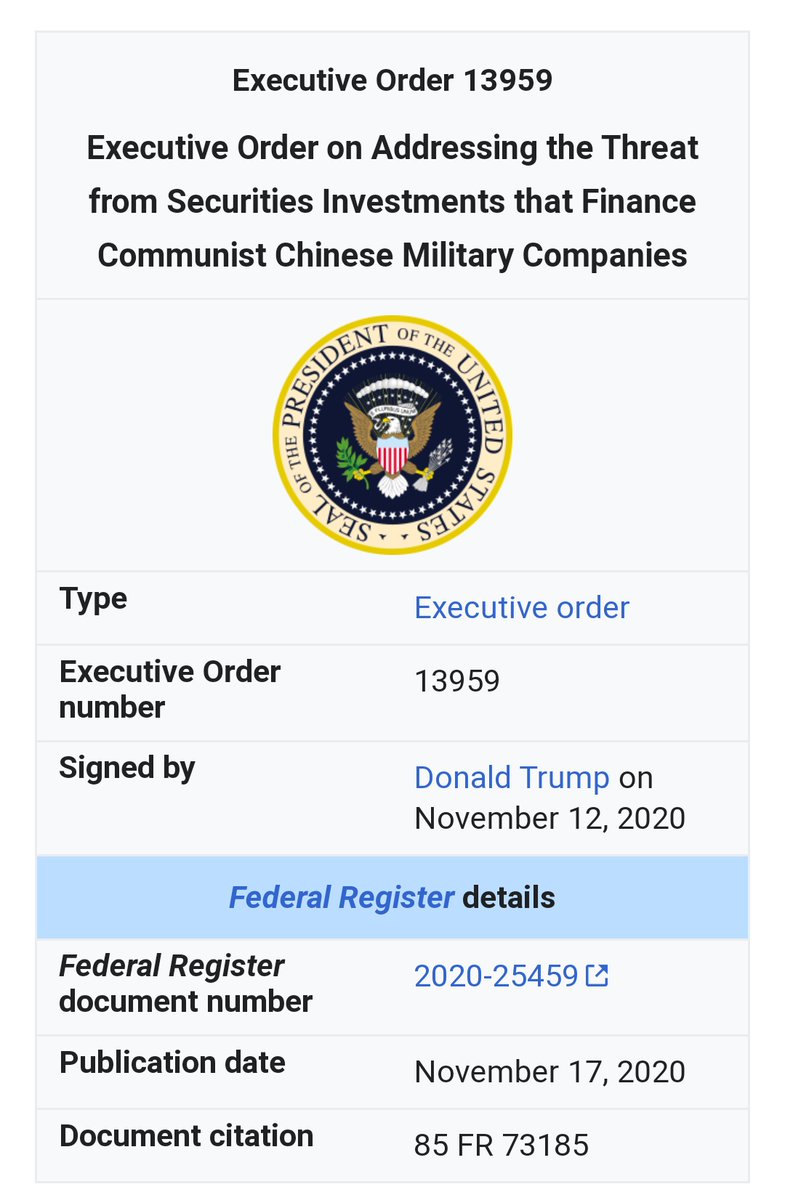
#ProjectOdin #Starlink #ElonMusk #QuantumInternet
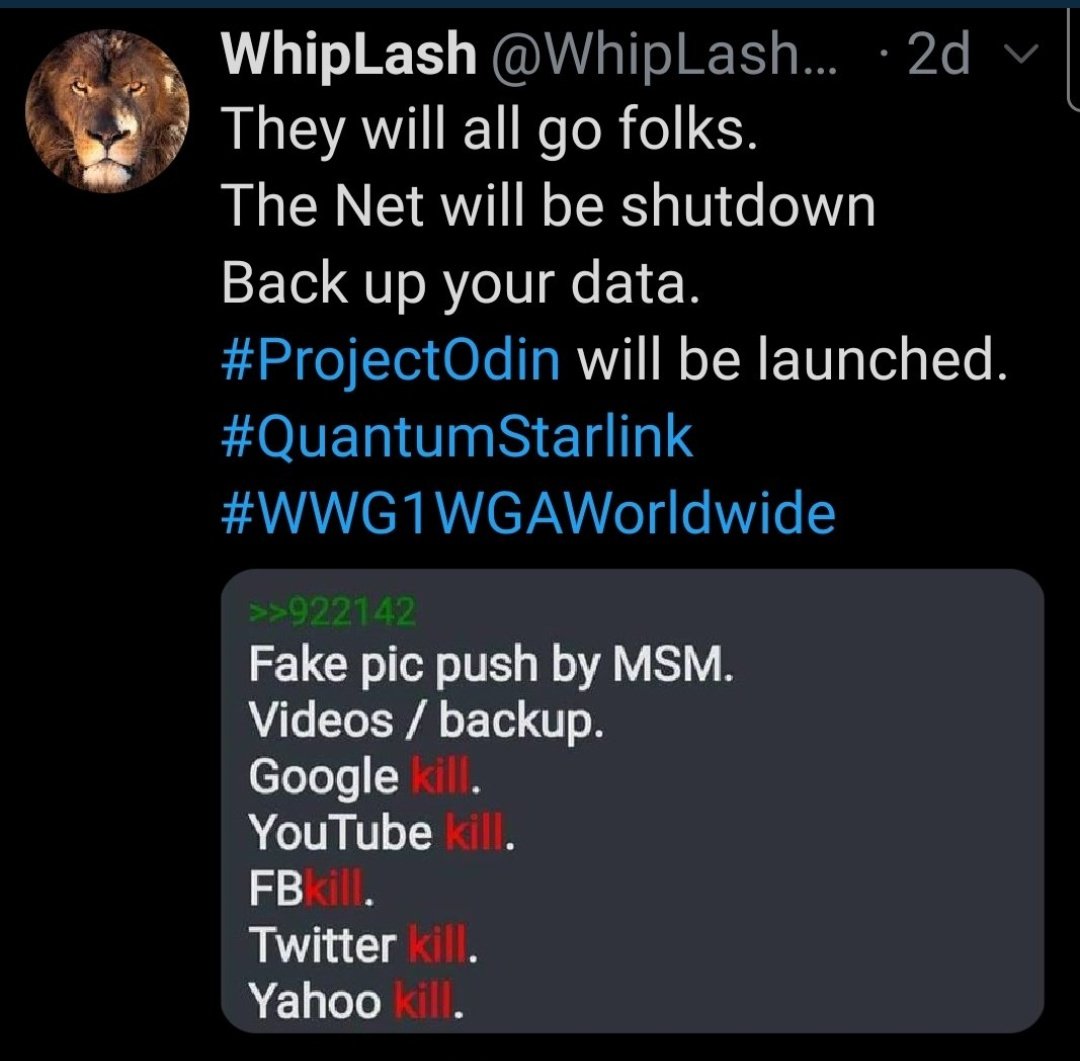
It's time, my friends 🤩🤩
[Thread] #ProjectOdin
The Alliance has Project Odin ready to go - the new quantum-based internet. #ElonMusk #QVS #QFS #ProjectOdin
— Der Preu\xdfe Parler: @DerPreusse (@DerPreusse1963) January 12, 2021
https://t.co/fO90N78fta
new quantum-based internet #ElonMusk #QVS #QFS
Political justification ⏬⏬
#ProjectOdin
#ProjectOdin #Starlink #ElonMusk #QuantumInternet

Hugh Everett's birthday! Pioneer of the Many-Worlds Interpretation of quantum mechanics. Let us celebrate by thinking about ontological extravagance. I will do so by way of analogy, because I have found that everyone loves analogies and nobody ever willfully misconstrues them.
We look at the night sky and see photons arriving to us, emitted by distant stars. Let's contrast two different theories about how stars emit photons.
One theory says, we know how stars shine, and our equations predict that they emit photons roughly uniformly in all directions. Call this the "Many-Photons Interpretation" (MPI).
But! Others object. That is *so many photons*. Most of which we don't observe, and can't observe, since they're moving away at the speed of light. It's too ontologically extravagant to posit a huge number of unobservable things!
So they suggest a "Photon Collapse Interpretation." According to this theory, the photons emitted toward us actually exist. But photons that would be emitted in directions we will never observe simply collapse into utter non-existence.
The physicist Hugh Everett III was born #OTD in 1930. His \u201crelative state\u201d formulation of quantum mechanics, which we now call the \u201cMany Worlds Interpretation,\u201d was published in 1957. pic.twitter.com/ZqMsZcPJDG
— Robert McNees, the bastegod (@mcnees) November 11, 2020
We look at the night sky and see photons arriving to us, emitted by distant stars. Let's contrast two different theories about how stars emit photons.
One theory says, we know how stars shine, and our equations predict that they emit photons roughly uniformly in all directions. Call this the "Many-Photons Interpretation" (MPI).
But! Others object. That is *so many photons*. Most of which we don't observe, and can't observe, since they're moving away at the speed of light. It's too ontologically extravagant to posit a huge number of unobservable things!
So they suggest a "Photon Collapse Interpretation." According to this theory, the photons emitted toward us actually exist. But photons that would be emitted in directions we will never observe simply collapse into utter non-existence.



You May Also Like


Krugman is, of course, right about this. BUT, note that universities can do a lot to revitalize declining and rural regions.
See this thing that @lymanstoneky wrote:
And see this thing that I wrote:
And see this book that @JamesFallows wrote:
And see this other thing that I wrote:
One thing I've been noticing about responses to today's column is that many people still don't get how strong the forces behind regional divergence are, and how hard to reverse 1/ https://t.co/Ft2aH1NcQt
— Paul Krugman (@paulkrugman) November 20, 2018
See this thing that @lymanstoneky wrote:
And see this thing that I wrote:
And see this book that @JamesFallows wrote:
And see this other thing that I wrote:

Department List of UCAS-China PROFESSORs for ANSO, CSC and UCAS (fully or partial) Scholarship Acceptance
1) UCAS School of physical sciences Professor
https://t.co/9X8OheIvRw
2) UCAS School of mathematical sciences Professor
3) UCAS School of nuclear sciences and technology
https://t.co/nQH8JnewcJ
4) UCAS School of astronomy and space sciences
https://t.co/7Ikc6CuKHZ
5) UCAS School of engineering
6) Geotechnical Engineering Teaching and Research Office
https://t.co/jBCJW7UKlQ
7) Multi-scale Mechanics Teaching and Research Section
https://t.co/eqfQnX1LEQ
😎 Microgravity Science Teaching and Research
9) High temperature gas dynamics teaching and research section
https://t.co/tVIdKgTPl3
10) Department of Biomechanics and Medical Engineering
https://t.co/ubW4xhZY2R
11) Ocean Engineering Teaching and Research
12) Department of Dynamics and Advanced Manufacturing
https://t.co/42BKXEugGv
13) Refrigeration and Cryogenic Engineering Teaching and Research Office
https://t.co/pZdUXFTvw3
14) Power Machinery and Engineering Teaching and Research
1) UCAS School of physical sciences Professor
https://t.co/9X8OheIvRw
2) UCAS School of mathematical sciences Professor
3) UCAS School of nuclear sciences and technology
https://t.co/nQH8JnewcJ
4) UCAS School of astronomy and space sciences
https://t.co/7Ikc6CuKHZ
5) UCAS School of engineering
6) Geotechnical Engineering Teaching and Research Office
https://t.co/jBCJW7UKlQ
7) Multi-scale Mechanics Teaching and Research Section
https://t.co/eqfQnX1LEQ
😎 Microgravity Science Teaching and Research
9) High temperature gas dynamics teaching and research section
https://t.co/tVIdKgTPl3
10) Department of Biomechanics and Medical Engineering
https://t.co/ubW4xhZY2R
11) Ocean Engineering Teaching and Research
12) Department of Dynamics and Advanced Manufacturing
https://t.co/42BKXEugGv
13) Refrigeration and Cryogenic Engineering Teaching and Research Office
https://t.co/pZdUXFTvw3
14) Power Machinery and Engineering Teaching and Research






