Authors Bloom Lab
7 days
30 days
All time
Recent
Popular

I'm agnostic on its various hypotheses about mechanisms of origin of furin-cleavage sites, but the part of this paper that suggests furin-cleavage site might be present in two of these SARSr-CoVs as a minor variant is embarrassingly bad science that shouldn't be amplified. (1/n)
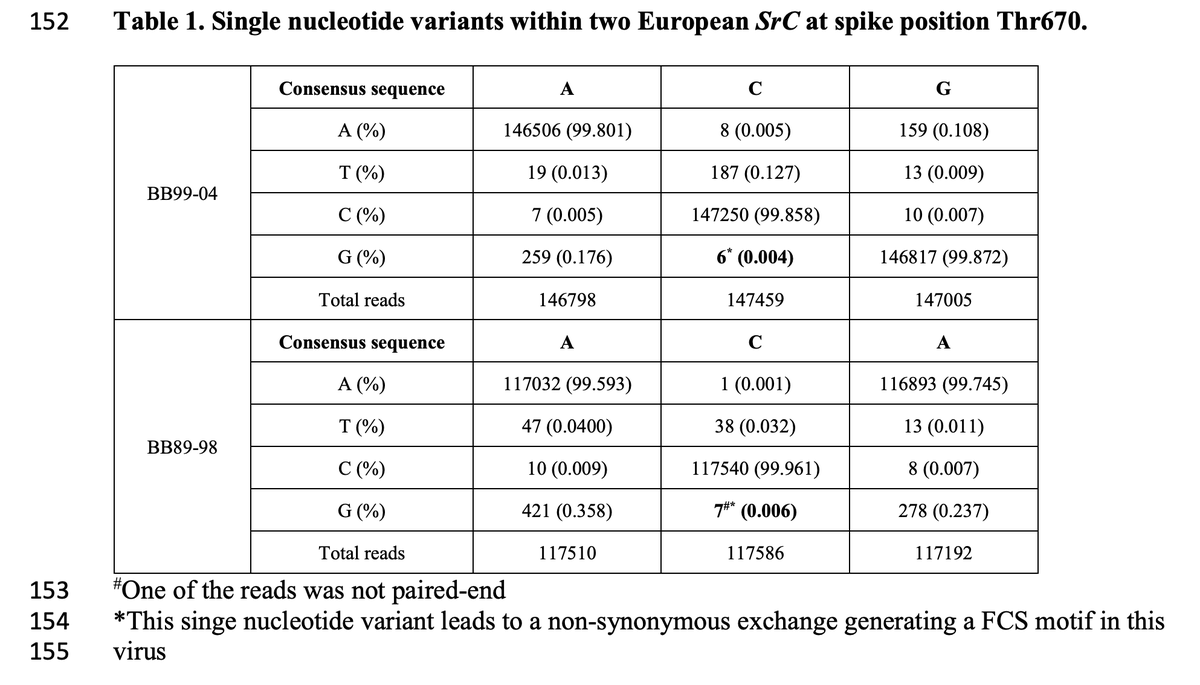
Here is Table 1 of pre-print (https://t.co/7fQsreN77n) that shows data related to this claim. The mutations in question are present at 0.004% and 0.006%, corresponding to 6 or 7 Illumina reads out of >100,000 total. (2/n)
There are good deep-sequencing studies of CoVs, eg by @katrina_lythgoe (https://t.co/joX4kCqOEh), @LauringLab (https://t.co/TisAPvkCVp) & @KATarinambraun @tcfriedrich @trvrb @LouiseHMoncla (https://t.co/08EhfVAAL1). These studies find you can call variants to 2-3% frequency (3/n)

Table 1 of this pre-print is reporting "mutations" that introduce a furin-cleavage site at frequencies of 0.004% to 0.006%, which is orders of magnitude lower than what good studies have rigorously defined as a reasonable threshold (2-3%) to call mutations. (4/n)
As anybody who has ever analyzed viral or cancer deep sequencing knows, it's simply impossible to use standard Illumina sequencing to identify mutations at even 0.4% or 0.04%, let alone 0.004%. It's actually surprising when a given sequencing error isn't present at ~0.01%. (5/n)
Back to CoVs. In samples from two European bats, the authors found SARSr-CoVs that were just one mutation away from a functional FCS. But minor variants were sequenced from each that *had* a functional FCS already, just as is seen with some low path flu.
— Michael Worobey (@MichaelWorobey) December 16, 2021
11/
Here is Table 1 of pre-print (https://t.co/7fQsreN77n) that shows data related to this claim. The mutations in question are present at 0.004% and 0.006%, corresponding to 6 or 7 Illumina reads out of >100,000 total. (2/n)
There are good deep-sequencing studies of CoVs, eg by @katrina_lythgoe (https://t.co/joX4kCqOEh), @LauringLab (https://t.co/TisAPvkCVp) & @KATarinambraun @tcfriedrich @trvrb @LouiseHMoncla (https://t.co/08EhfVAAL1). These studies find you can call variants to 2-3% frequency (3/n)
Table 1 of this pre-print is reporting "mutations" that introduce a furin-cleavage site at frequencies of 0.004% to 0.006%, which is orders of magnitude lower than what good studies have rigorously defined as a reasonable threshold (2-3%) to call mutations. (4/n)
As anybody who has ever analyzed viral or cancer deep sequencing knows, it's simply impossible to use standard Illumina sequencing to identify mutations at even 0.4% or 0.04%, let alone 0.004%. It's actually surprising when a given sequencing error isn't present at ~0.01%. (5/n)